Nouvelles connaissances sur l'acidité de Lewis des espèces de guanidinium: l'interaction acide de Lewis fournit la réactivité
Comptes Rendus. Chimie, 2020, 23, no. 2, p. 185-199
Muhammad Ageel Ashraf ; Cheng Li ; Fataneh Norouzi ; Dangquan Zhang
(traduction post-éditée par N. Bacaër. Suggestions d'amélioration : nicolas.bacaer@ird.fr)
Résumé
Un modèle de réactivité unique dans lequel le catalyseur et les motifs réactifs sont réunis dans un substrat est étudié en utilisant des calculs de théorie fonctionnelle de densité. La fonctionnalité polyvalente triaza CN3 dans les guanidines bicycliques est combinée avec une diversité de groupes fonctionnels sur l'échafaudage moléculaire qui, lors de l'addition de N-nucléophiles, transfère le substrat à ses analogues tricycliques non signalés jusqu'à présent. Sans précédent, la dyade guanidine / guanidinium fait progresser la réaction en affichant toutes les fonctionnalités chimiques possibles, y compris les modes d'activation acide-base de Brønsted et acide-base de Lewis. Cette étude présente le premier exemple d'une réaction de thioamidation catalysée par la guanidine. De plus, il met en évidence la première participation de l'activation de l'acide de Lewis du guanidinium dans une réaction de cyclisation. En plus des différents modes d'activation mono-et bifonctionnels, deux rares exemples d'interaction acide de Lewis ont été observés, ce qui fournit des points mécanistes importants. Le premier dans l'étape de thioamidation suggère la préférence pour l'élimination par étapes de MeSH sur les mécanismes concertés alternatifs. Cependant, le second dans l'étape d'addition-N de Michael fournit un nouvel aperçu du mode d'activation de l'acidité de Lewis des espèces de guanidinium. L'interaction piège d'abord la branche latérale du substrat contenant le motif nucléophile, puis, en positionnant le nucléophile anionique et le site accepteur de Michael à proximité immédiate, dirige l' addition N de Michael régiosélective .
1. Introduction
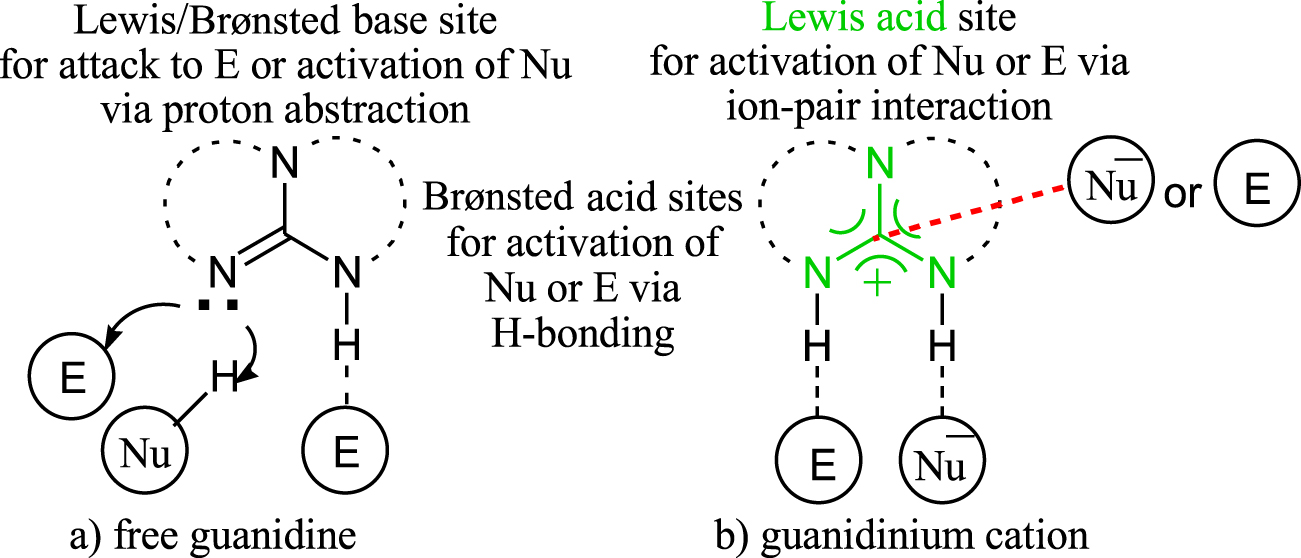
Les guanidines, en raison de leur richesse en fonctionnalités catalytiques, occupent une place de choix dans le répertoire des organocatalyseurs [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12]. Ils remplissent les quatre fonctions chimiques de base: les guanidines libres sont capables d'agir comme des bases de Brønsted ou de Lewis, tandis que leurs acides conjugués correspondants (cation guanidinium) peuvent servir d'acides de Brønsted ou de Lewis (schéma 1 ). Ces modes d'activation permettent au triaza (CN3) d'être un outil efficace dans les transformations organiques, telles que la réaction de Michael [13, 14, 15, 16], la réaction de Mannich [17, 18, 19, 20], la réaction d'aldol [21, 22, 23], l'ouverture de cycle polymérisation [24, 25, 26, 27, 28], réaction d'époxydation [29, 30, 31, 32] et plein d'autres. Les guanidines sont connues depuis longtemps comme des bases organiques fortes [33].
Schéma 1. Représentation schématique des fonctionnalités dans (a) guanidine libre et (b) cation guanidinium.
Cependant, les connaissances sur la caractéristique d'acidité de Lewis du cation guanidinium sont rares. Le cation guanidinium est produit lorsqu'une guanidine libre extrait un proton d'un substrat via le mécanisme de base de Brønsted (schéma 1 b). Grâce à des études soutenues par RMN, Olah et ses collègues ont démontré que l'aromaticité Y du système CN3 fournit une force motrice considérable pour la déprotonation des échafaudages de guanidine. L'ensemble du processus aboutit à un sel de guanidinium, comportant un complexe base-acide de Lewis formé par interaction électrostatique entre l'atome de carbone central du CN3(Acide de Lewis) et l'électron nucléophile anionique ou non liant d'un électrophile (base de Lewis). Le centre acide de Lewis chargé positivement polarisera les partenaires ligaturés, ce qui conduit à l'amélioration de leur réactivité intrinsèque. Ce processus, appelé «catalyse acide de Lewis organique», distingue les charpentes guanidines des composés hétérocycliques car le terme «catalyse acide de Lewis» est presque exclusivement utilisé pour les sels métalliques ou les complexes métalliques [34]. Des études expérimentales et informatiques ont démontré que l'interaction de l'acide de Lewis empêche non seulement la désactivation, mais améliore également la nucléophilie d'une base de Lewis en augmentant la densité de la charge négative sur elle en tirant la densité électronique de l'acide de Lewis [35, 37].
Il a été affirmé que le cation guanidinium sert de catalyseur acide de Lewis dans quelques processus chimiques tels que la réaction d'ouverture de cycle [24, 37], le clivage du phosphodiester et de l'ADN [38] et la décomposition du formiate d'alkyle [39, 40]. Cependant, ces rapports ne sont pas étayés par des preuves théoriques. Le calcul de la théorie fonctionnelle de la densité (DFT) a été reconnu comme une méthode fiable pour éclairer ce mécanisme et la qualité des modes d'activation dans les réactions d'organocatalyseurs [41]. Cependant, l'enquête informatique n'a été menée que dans une poignée de cas où les espèces de guanidinium contribuent à la réaction via le mode d'activation de l'acide de Lewis (Scheme 2) [16, 42, 43, 44, 45, 46, 47, 48, 49, 50].
Cette lacune de la littérature a été attribuée aux difficultés de mise en place d'un support théorique. Ainsi, en 2012, Wong et ses collaborateurs ont rapporté une étude pionnière sur la caractérisation du mode d'activation acide bifonctionnel de Lewis – Brønsted (schéma 2 a) sous l'activité catalytique de la guanidine bicyclique 1,4,6-triazabicyclo [3.3.0] oct-4-ène (TBO) [16].
Le calcul a révélé la complexation acide-base de Lewis entre le guanidinium et le thiolate au cours de la réaction asymétrique thio-Michael. Plus tard, le même groupe de chercheurs a étendu son étude à une gamme plus large de substrats [42, 43, 44, 45, 46, 47, 48, 49]. En 2015, le groupe Wong a démontré que le mode d'activation acide bifonctionnel non conventionnel de Brønsted – Lewis remplissait une fonction catalytique dans la réaction d'isomérisation de l'alk-3-ynoate (schéma 2 b) [50].
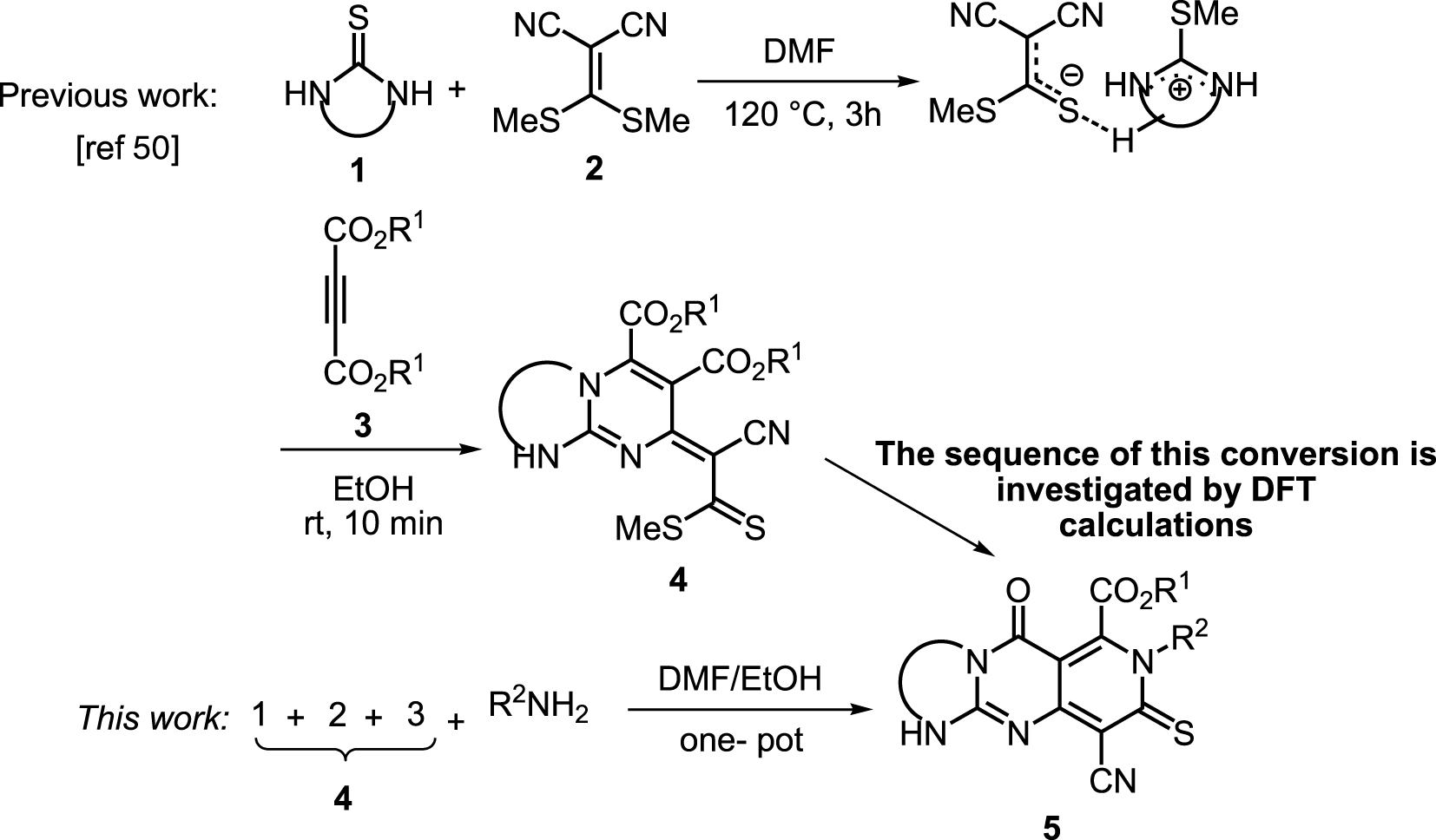
Ici, nous rapportons une réaction à quatre composants en un seul pot conduisant à la synthèse de guanidine tricyclique, contenant le noyau hétérocyclique sans précédent, à savoir la pyrido [4,3-d] pyrimido [1,2-a] pyrimidine (schéma 2 c) . Des calculs DFT ont été effectués pour étudier le mécanisme et les modes d'activation du groupe fonctionnel CN3 dans la guanidine bicyclique (précurseur). Parmi les différents modes d'activation, le rôle du récepteur anionique dans les interactions acide de Lewis ressort. Le calcul a révélé que la complexation acide-base de Lewis entre l'atome de carbone central fortement chargé positivement et l'atome de soufre anionique joue un rôle important dans les étapes de thioamidation et d' addition de N-Michael.
2. Expériences
2.1. Informations générales
Les points de fusion ont été mesurés sur un appareil électrothermique 9100. Les spectres infrarouges (IR) ont été enregistrés sous forme de pastilles KBr sur un spectromètre NICOLET FT-IR 100. Les spectres RMN 1 H (300 et 500 MHz) et RMN 13 C (75 et 100 MHz) ont été obtenus en utilisant les spectromètres Bruker DRX-300 AVANCE et Bruker DRX-500 AVANCE. Tous les spectres de résonance magnétique nucléaire (RMN) ont été enregistrés dans du DMSO-d 6 à la température ambiante. Les déplacements chimiques sont indiqués en parties par million ( δ) en aval d'une référence tétraméthylsilane interne. Les constantes de couplage (valeurs J) sont exprimées en hertz (Hz) et les multiplicités de spin sont indiquées par les symboles suivants: s (singulet), d (doublet), t (triplet), q (quartet) et m (multiplet). Des analyses élémentaires de C, H et N (CHN) ont été effectuées à l'aide d'un analyseur Heraeus CHN – O – Rapid. Les spectres de masse ont été enregistrés sur un spectromètre de masse FINNIGAN-MATT 8430 fonctionnant à un potentiel d'ionisation de 70 eV. Les coordonnées cartésiennes de toutes les structures optimisées sont jointes séparément sous forme de fichiers .xyz.
Schéma 2. Acidité de Lewis des cations guanidinium.
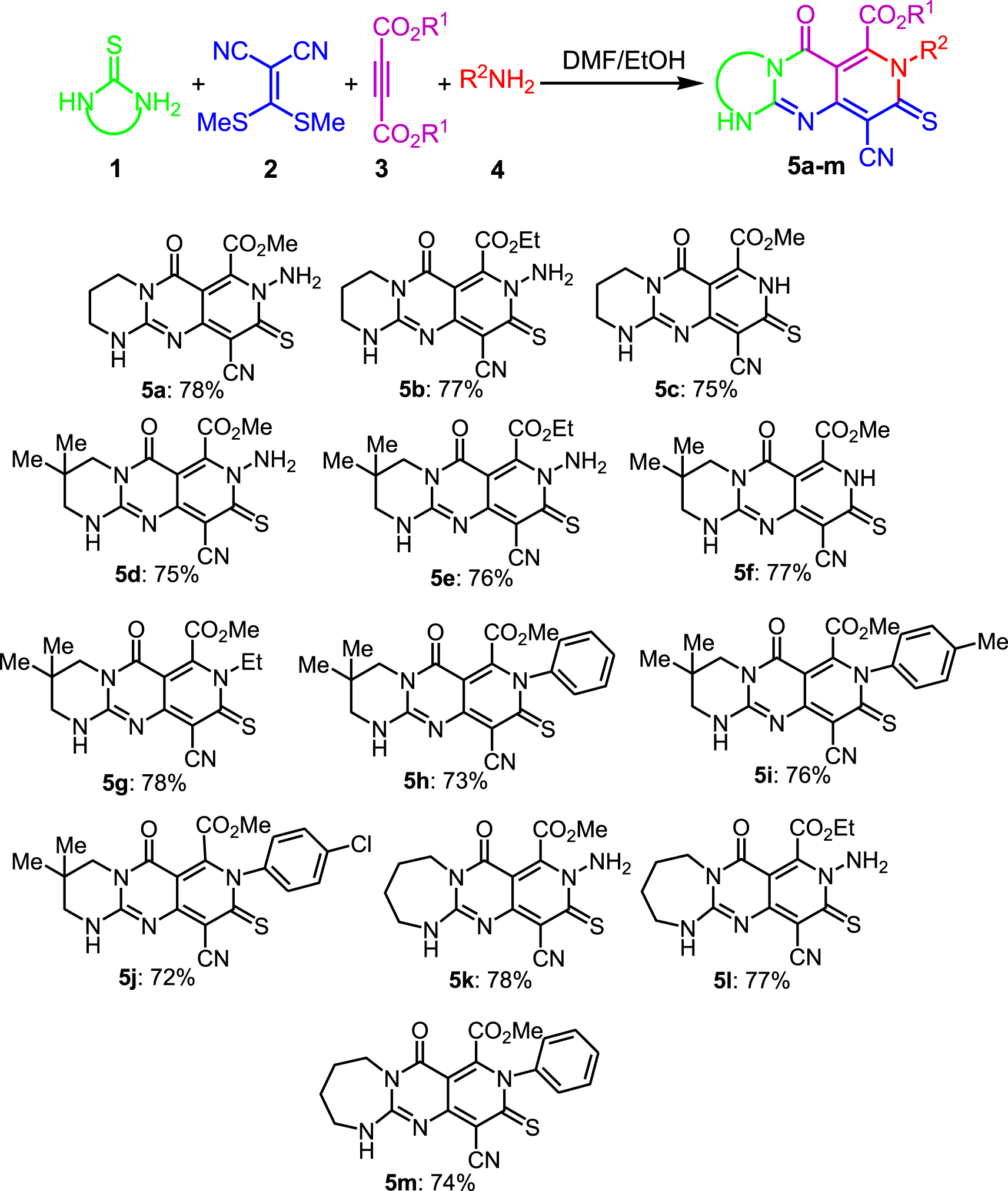
2.2. Procédure générale pour la synthèse des composés 5a-m
A une solution de la thiourée cyclique correspondante 1 (1 mmol) dans du DMF (1,0 ml), a été ajouté du 2-di (méthylsulfanyl) méthylène-malononitrile 2 (0,170 g, 1 mmol). Le mélange réactionnel a été agité pendant 3 h à 120 °C. Ensuite, une solution d'acétylènedicarboxylates de dialkyle 3 (1 mmol) dans EtOH (2,0 ml) a été ajoutée goutte à goutte au mélange réactionnel à température ambiante. Une fois l'addition terminée, l'agitation s'est poursuivie pendant encore 20 minutes. Ensuite, les N- nucléophiles 4 correspondants (1,2 éq) ont été ajoutés au mélange. Après agitation pendant 1 h à température ambiante, le solvant a été évaporé par un évaporateur rotatif et le résidu a été lavé par EtOH pour donner les produits d'addition souhaités 5a-m .
Méthyle 8-(4-chlorophényl)-10-cyano-3,3-diméthyl-6-oxo-9-thioxo-1,3,4,6,8,9-hexahydro-2H-pyrido [4,3-d] pyrimido [1,2-a] pyrimidine-7-carboxylate ( 5j ). Poudre jaune, point de fusion> 300 °C, 0,328 g, rendement: 72%. IR (KBr) ( νmax, cm-1 ): 3210 (NH), 2216 (CN), 1747 (CO2 Me), 1690 (C = O), 1605 (C = N), 15777 (C = C), 1295 (C = S), 1080 (C – O). Anal. Calcd. pour C21 H18 ClN5 O3 S (455,92): C, 55,32; H, 3,98; N, 7,78%. Trouvé C, 55,29; H, 4,01; N, 7,77. RMN1H (300 MHz, DMSO-d6 ): 1,03 (6H, s, CMe2 ), 3,12 (2H, m, CH2N), 3,50 (3H, s, CO2 Me), 3,58 (2H, m, CH2 NH), 7,13 à 7,62 (4H, m, 4CH de Ar), 9,60 (1H, s large, NH).13C {1H} (100,0 MHz, DMSO-d6 ): 23,69, 27,09, 50,13, 53,73, 103,87, 104,37, 117,01, 129,38, 129,49, 134,91, 138,42, 147,41, 154,19, 157,38, 159,11, 161,25, 181,60. SM (EI, 70 eV): 410 (28), 380 (34), 320 (45), 284 (27), 254 (66), 223 (44), 169 (37), 111 (100), 73 ( 29).
Méthyle 9-amino-11-cyano-7-oxo-10-thioxo-1,2,3, 4,5,7,9,10 octahydropyrido [4 ', 3': 4,5] pyrimido [1,2-a] [1,3] diazépine-8-carboxylate ( 5k ). Poudre jaune, point de fusion> 300 °C, 0,270 g, rendement: 78%. IR (KBr) ( νmax, cm-1 ): 3211 et 3118 (NH2 et NH), 2223 (CN), 1750 (CO2 Me), 1692 (C = O), 1610 (C = N), 1571 (C = C), 1295 (C = S), 1064 (C-O). Anal. Calcd. pour C14 H14 N6 O3 S (346,36): C, 48,55; H, 4,07; N, 24,46%. Trouvé C, 48,56; H, 4,09; N, 24,46. RMN1H (300 MHz, DMSO-d6 ): 1,86 (4H, m, 2CH2 ), 3,43–3,47 (2H, m, CH2 N), 3,91 (3H, s, CO2 Me), 4.5 à 4.9 (2H, m, CH2 NH), 6,57 (2H, s, NH2 ), 9,58 (1H, s large, NH).13C {1H} (75,0 MHz, DMSO-d6 ): 23,96, 24,57, 42,53, 42,59, 53,83, 103,13, 103,31, 116,90, 147,13, 156,40, 159,68, 159,93, 160,73, 179,76. SM (EI, 70 eV): 345 (M+-1, 15), 316 (22), 279 (34), 241 (31), 216 (29), 191 (42), 149 (40), 111 ( 57), 73 (100), 44 (89). Données cristallines pour 5k C14 H14 N6 O3 S, C2 H3 N1 (CCDC 1551358): MW= 387,44, monoclinique, P21 / n, 11,5060 (13) Å, b = 9,5212 (12) Å, c = 16,697 (2) Å, 𝛼 = 90,00, 𝛽 = 100,160 (10), 𝛾 = 90,00, V = 1800,5 ( 4) Å3, Z = 4, Dc = 1,429 mg / m3, F (000) = 808, MoK α ( λ = 0,71073 Å). Les données d'intensité ont été recueillies à 298 (2) K avec un diffractomètre STOE IPDS-II avec monochromateur en graphite et en utilisant la technique de balayage 𝜔 ∕ 2𝜃 dans la plage de-15⩽ h ⩽15,-12⩽ k ⩽13,-22⩽ l⩽22. La structure a été résolue par une méthode directe; tous les atomes non hydrogène ont été positionnés et les paramètres thermiques anisotropes ont été affinés à partir de 4881 réflexions observées avec R (int) = 0,2409 par une technique des moindres carrés à matrice complète convergeant vers R = 0,0679 et w R2 = 0,0913 [I> 2sigma (I )].
Éthyl 9-amino-11-cyano-7-oxo-10-thioxo-1,2,3,4, 5,7,9,10 octahydropyrido [4 ', 3': 4,5] pyrimido [1,2-un] [1,3] diazépine-8-carboxylate d' éthyle ( 5l ). Poudre jaune, point de fusion> 300 °C, 0,277 g, rendement: 77%. IR (KBr) ( νmax, cm-1 ): 3211 et 3118 (NH2 et NH), 2223 (CN), 1750 (CO2 Me), 1692 (C = O), 1610 (C = N), 1571 (C = C), 1295 (C = S), 1088 (C-O). Anal. Calcd. pour C15 H16 N6 O3 S (360,40): C, 49,99; H, 4,47; N, 23,32%. Trouvé C, 50,02; H, 4,44; N, 23h30. RMN1H (300 MHz, DMSO-d6 ): 1,32-1,36 (3H, m, OCH2CH3 ), 1,89–1,91 (4H, m, 2CH2 ), 3,43–3,47 (2H, m, CH2 N), 4,08–4,12 (2H, m, CH2 NH), 4,40–4,42 (2H, m, OCH2 CH3 ), 6,60 (2H, s, NH2 ), 8,84 (1H, s large, NH).13C {1H} (75,0 MHz, DMSO-d6 ): 14,08, 23,98, 24,58, 42,58, 63,11, 103,01, 103,26, 116,95, 147,72, 156,41, 159,67, 159,92, 160,14, 179,70. SM (El, 70 eV): 360 (M+, 31), 328 (42), 299 (100), 256 (32), 227 (49), 186 (38), 132 (22), 97 (64), 54 (62).
Récemment, Alizadeh et al. ont rapporté une réaction en deux étapes pour la synthèse de guanidines bicycliques 4 hautement fonctionnalisées en utilisant la thiourée cyclique 1, le cétène dithioacétal 2 et les acétylènedicarboxylates de dialkyle 3 (schéma 3) [51].
Schéma 3. Conversion des guanidines bicycliques en guanidines tricycliques.
En raison des avantages des réactions à un seul pot, nous avons commencé l'étude actuelle en ajoutant les adduits bicycliques 4 à un processus en tandem en un seul pot. Ensuite, l'addition de N- nucléophiles au mélange de cette réaction, contenant les guanidines bicycliques générées in situ, a conduit aux adduits tricycliques de guaïdine 5a-m avec de bons rendements (tableau 1).
Tableau 1.
Synthèse à quatre composants en un seul pot de guanidines tricycliques[a]
a Conditions de réaction: un pot, 1 (1 mmol), 2 (1 mmol), 2 mL de DMF, 120 °C, 3 h; 3 suivants (1 mmol), 2 ml d'EtOH, rt, 1 h, 4 suivants (1,2 mmol), rt, 1h.
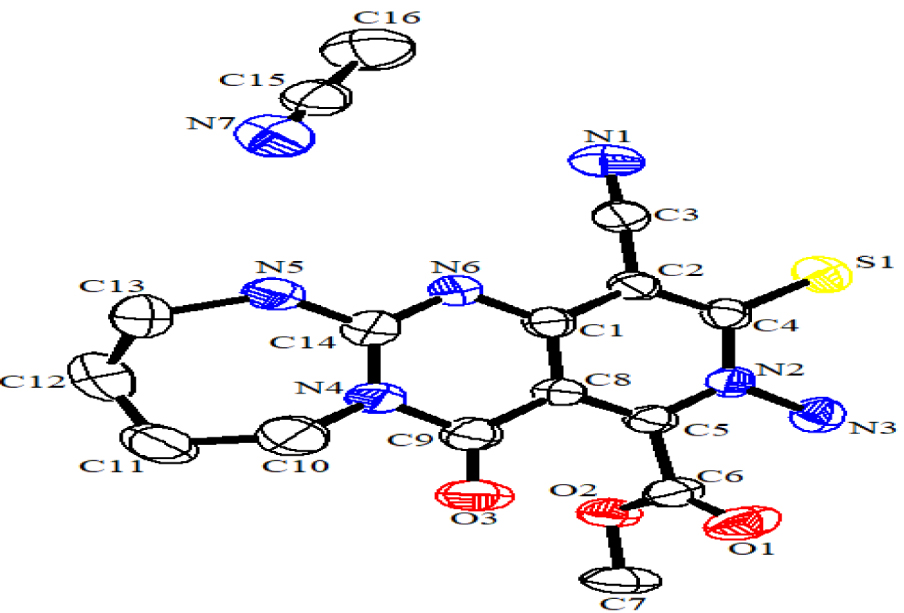
Nous avons constaté que l'efficacité de la procédure n'est pas sensible à l'électronique des précurseurs. De plus, d'autres expériences ont démontré que la présence d'un solvant alcoolique et la quantité excessive de nucléophile (1,2 équivalent) sont critiques pour la transformation finale (conversion de la guanidine bicyclique en guanidine tricyclique). Hormis les analyses CHN, IR, masse, RMN1H et RMN13C pour tous les composés, la structure de 5k comme exemple représentatif est en outre confirmée par cristallographie aux rayons X (schéma 4 ).
Schéma 4. Diagramme ORTEP de 5k .
3.2. Méthodes de calcul
Pour étudier l'origine de cette transformation, le diméthyl 2-[(Z)-1-cyano-2-(méthylsulfanyl)-2-thioxoéthylidène]-6,7,8,9-tétrahydro-2H-pyrimido [1, Le 2-a] pyrimidine-3,4-dicarboxylate et l'hydrazine (conduisent à 5a dans le tableau 1) ont été sélectionnés comme réaction modèle. L'investigation informatique de la réaction est classée en deux sections principales comprenant la thioamidation et l' addition de N- Michael. Il a été démontré que M06−2X est une fonction empirique efficace pour étudier les interactions non covalentes, y compris les interactions zwitterioniques à longue portée dans les réactions catalysées par le guanidinium [16, 49, 52, 53]. Par conséquent, toutes les géométries des complexes réactifs (RC), des intermédiaires (INT), des états de transition (TS), des complexes de produits (PrC) et des produits (Prs) ont été optimisées au niveau de théorie M06−2X en conjonction avec le 6-31G * ensemble de base. La coordonnée de réaction intrinsèque (IRC) a été effectuée pour vérifier tous les TS. Le modèle de solvatation basé sur la densité (SMD) [54] a été appliqué sur les géométries optimisées par le calcul en un point M06−2X / 6-311 + G ** pour examiner l'effet de solvatation de l'éthanol. La densité de charge sur les atomes a été calculée par analyse orbitale de liaison naturelle (NBO) [55] basée sur la méthode M06−2X / 6-31G * en phase gazeuse. Toutes les optimisations ont été réalisées à l'aide du logiciel Orca 4 [56">56].
3.2.1. Étape de thioamidation
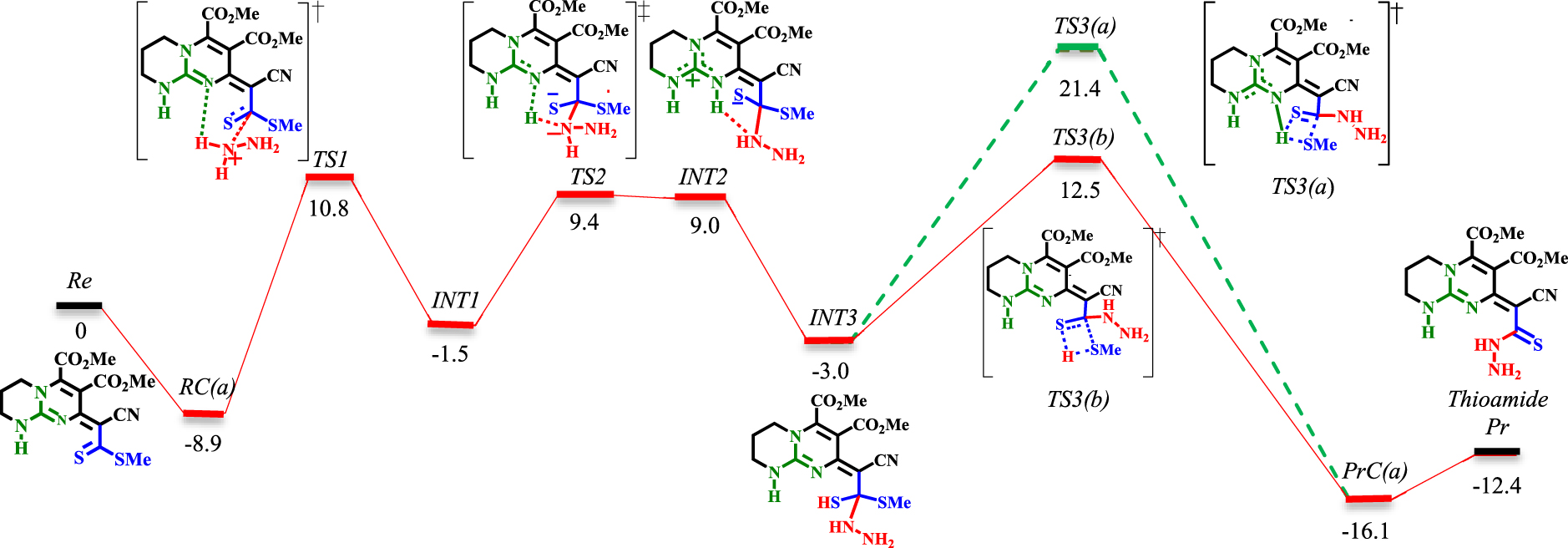
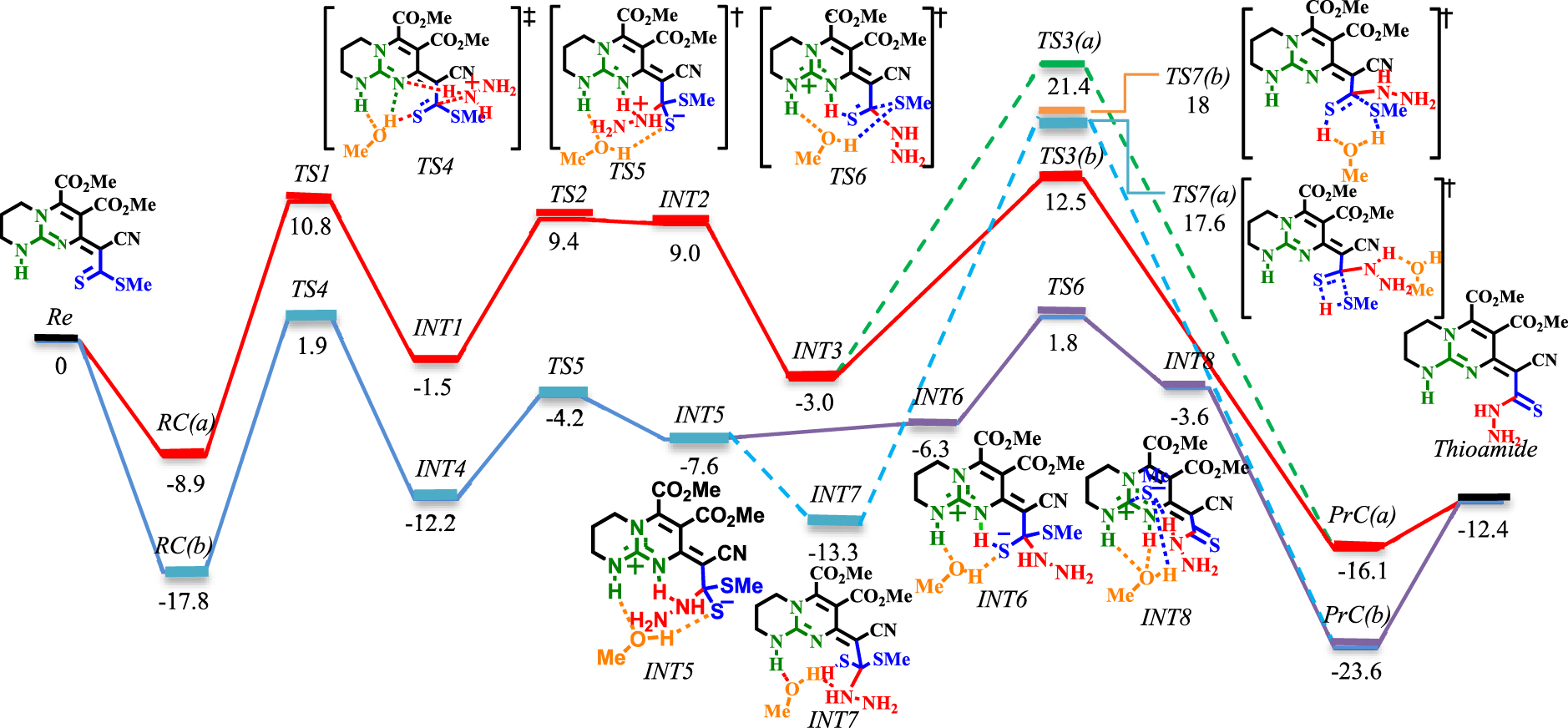
Schéma 5. Profil énergétique de l'étape de thioamidation. Les valeurs sont des énergies relatives (𝛥 E298 dans le solvant éthanol, kcal⋅mol−1 ) calculées au niveau M06−2X / 6-311 + G ** // M06-2X / 6-31G *.Schéma 6. Profil énergétique de l'étape de thioamidation sous solvatation explicite de MeOH (lignes bleues et violettes). Les valeurs sont des énergies relatives (𝛥 E298 dans le solvant éthanol, kcal⋅mol−1 ) calculées au niveau M06−2X / 6-311 + G ** // M06−2X / 6-31G *.
La réaction est étudiée via deux approches principales (voir schémas 5 et 6). Dans le premier, seules la guanidine et l'hydrazine bicycliques sont considérées (lignes rouges et vertes dans les schémas 5 et 6). Dans le second, le méthanol (pour simplifier les calculs, MeOH est remplacé par EtOH) est explicitement inclus comme solvant (lignes bleues et violettes dans le schéma 6). Par souci de simplicité, seules les structures des TS et certaines des INT importantes sont données (une séquence complète de réaction impliquant les structures de toutes les INT peut être trouvée dans les données supplémentaires).
La réaction commence avec le complexe du réactif bimoléculaire RC (a), qui est ensuite procédé par formation de liaison C-N ([TS1] ‡). Cette étape (RC (a) → [TS1] ‡ → INT1) nécessite de franchir une barrière d'énergie de 19,7 kcal⋅mol−1 . Impliquer du méthanol comme solvant nécessite la même quantité d'énergie d'activation (RC (b) → [TS4] ‡ → INT4). Cependant, la présence d'un solvant donneur de liaison H réduit significativement les surfaces d'énergie de RC (b), [TS4] ‡, INT4 par rapport à RC (a), [TS1] ‡, INT1. L'étape suivante est la transformation du proton d'un ion ammonium en un groupe imine par un mécanisme à base de Brønsted. Cette étape a une barrière de 10,9 kcal⋅mol−1 (INT1 → [TS2] ‡ → INT2). Cependant, lorsque le méthanol est impliqué, la barrière est abaissée à 8 kcal⋅mol-1(INT4 → [TS5] ‡ → INT5). L'élimination du méthanethiol (MeSH) est la dernière étape du processus de thioamidation. Dans les deux conditions, le proton est transféré de l'acide de Brønsted (NH du guanidinium) au thiolate (INT2 → INT3 et INT5 → INT7). L'élimination de MeSH pourrait se faire via [TS3 (a)] ‡ ou [TS3 (b)] ‡ avec des barrières de 24,4 et 15,5 kcal⋅mol−1, respectivement (via le mécanisme de décalage 1,3-H). En revanche, en procédant de [TS7 (a)] ‡ (via le mécanisme de décalage 1,3-H) ou [TS7 (b)] ‡ (via le mécanisme de navette à protons), des barrières plus élevées (30,9 kcal⋅mol−1 pour [TS7 (a)] ‡ et 31,3 kcal⋅mol−1 pour [TS7 (b)] ‡) sont rencontrés. En raison de l'implication de barrières supérieures à 30 kcal⋅mol−1, la possibilité de partir de [TS7 (a)] ‡ et [TS7 (b)] ‡ diminue. Heureusement, l'étude d'une voie alternative (de INT5 au produit thioamide, ligne violette dans le schéma 6) via [TS6] ‡ a conduit à la réaction la plus favorable à travers une barrière de 9,4 kcal⋅mol−1 . Cette étape est exergonique avec une énergie de 4,8 kcal⋅mol−1 en référence à INT5. Ici, INT5 évoluera facilement vers INT6 avec une barrière de 1,3 kcal⋅mol−1 . INT6 subit alors un clivage de liaison C-SMe à la suite de l'assistance MeOH lors de l'isomérisation du thiolate en thiocarbonyle (8,1 kcal⋅mol-1 pour INT6 → [TS6] ‡).
Comme le montre le schéma 6, la barrière inférieure [TS6] ‡ est caractérisée par un réseau intramoléculaire en forme de chaîne lié en H entre le substrat polarisé et la molécule MeOH. Chaque partie de cette chaîne se concentre pour redistribuer les changements d'électrons sur les hétéroatomes impliqués, ce qui conduit à la tautomérisation et à la sortie du groupe MeS-. Cette coopération est similaire au mécanisme de «proton hopping», d'abord suggéré par Grotthuss [57]. L'espèce thion-thiolate est stabilisée par liaison H avec le NH du guanidinium (activation acide de Brønsted). Parallèlement, comme la liaison NH ⋯ S s'affaiblit, le clivage de la liaison C-SMe est encouragé par la liaison H intramoléculaire avec MeOH. Cela augmentera la densité électronique dans la région à paire unique de MeOH, ce qui persuade l'acceptation de la liaison H du deuxième NH du guanidinium. Ainsi, si les deux NH sont considérés comme les deux conducteurs de cette chaîne, l'affaiblissement du don H d'un côté est compensé par le renforcement du don de l'autre côté. Par conséquent, l'acidité de Brønsted bifonctionnelle du guanidinium en coopération avec MeOH génère la force motrice pour le clivage de la liaison C-SMe.
L'observation de INT8 (schéma 6) donne un aperçu instructif du mécanisme d'élimination de MeSH. Comme cela a été démontré en théorie, l'élimination progressive de MeSH via [TS6] ‡ est énergétiquement plus favorisée que les processus d'élimination concertée (à comparer avec [TS3 (a)] ‡, [TS3 (b)] ‡, [TS7 (a)] ‡ et [TS7 (b)] ‡). Cette découverte est contraire au mode de dissociation de la ROH dans la polymérisation par ouverture de cycle de la lactone [58] et la transestérification / amidation de l'ester aromatique [59]. Dans ces réactions, les résultats calculés ont démontré que le clivage de la liaison C-OR et de la transformation protonique est concerté. Pour valider davantage le complexe acide-base de Lewis dans INT8, des diagrammes de la structure optimisée, des orbitales moléculaires et de l'analyse NBO sont donnés dans les schémas S1 et S2 (voir données supplémentaires).
La géométrie optimisée de INT8 montre que le centre acide de Lewis du guanidinium penche vers le MeS-avec une distance d'interaction de 3,07 Å. De manière concomitante, MeS-forme une liaison H avec MeOH (2,11 Â). D'autre part, MeOH forme une liaison H bidentée (1,83 Â et 1,93 Â) avec les deux protons NH. L'ancienne liaison H renforce l'interaction entre l'acide de Lewis (CN3 ) et la base de Lewis (MeS-). Comme pour le cas de [TS6] ‡, la même discussion peut être effectuée pour INT8. La liaison H entre MeOH et MeS-augmente la densité électronique sur l'atome d'oxygène. Cela conduit à une augmentation de la capacité d'acceptation des liaisons H de MeOH à partir des groupes NH. Par conséquent, le carbone central du CN3devient plus déficient en électrons et un meilleur récepteur anionique pour MeS−. Par conséquent, un mode d'activation non conventionnel, à savoir l'activation acide bifonctionnelle de Lewis – Brønsted, est établi pour l'élimination du MeSH.
Dans l'ensemble, l'étape de thioamidation est exergonique de-12,4 kcal⋅mol-1, ce qui suggère qu'il existe une force motrice importante pour la formation d'un adduit de thioamide. Comme cela a été prouvé en théorie, l'inclusion explicite de MeOH affecte de manière significative l'étape de thioamidation. Il n'abaisse pas seulement les barrières d'activation mais affecte également la nature de l'étape de détermination de la vitesse (RDS) par rapport au mécanisme bimoléculaire. En conséquence, l'élimination de MeSH ([TS3 (b)] ‡) est le RDS dans le mécanisme bimoléculaire. En comparaison, la formation de liaison C − N ([TS4] ‡) correspond au RDS lorsque MeOH est impliqué comme troisième partenaire.
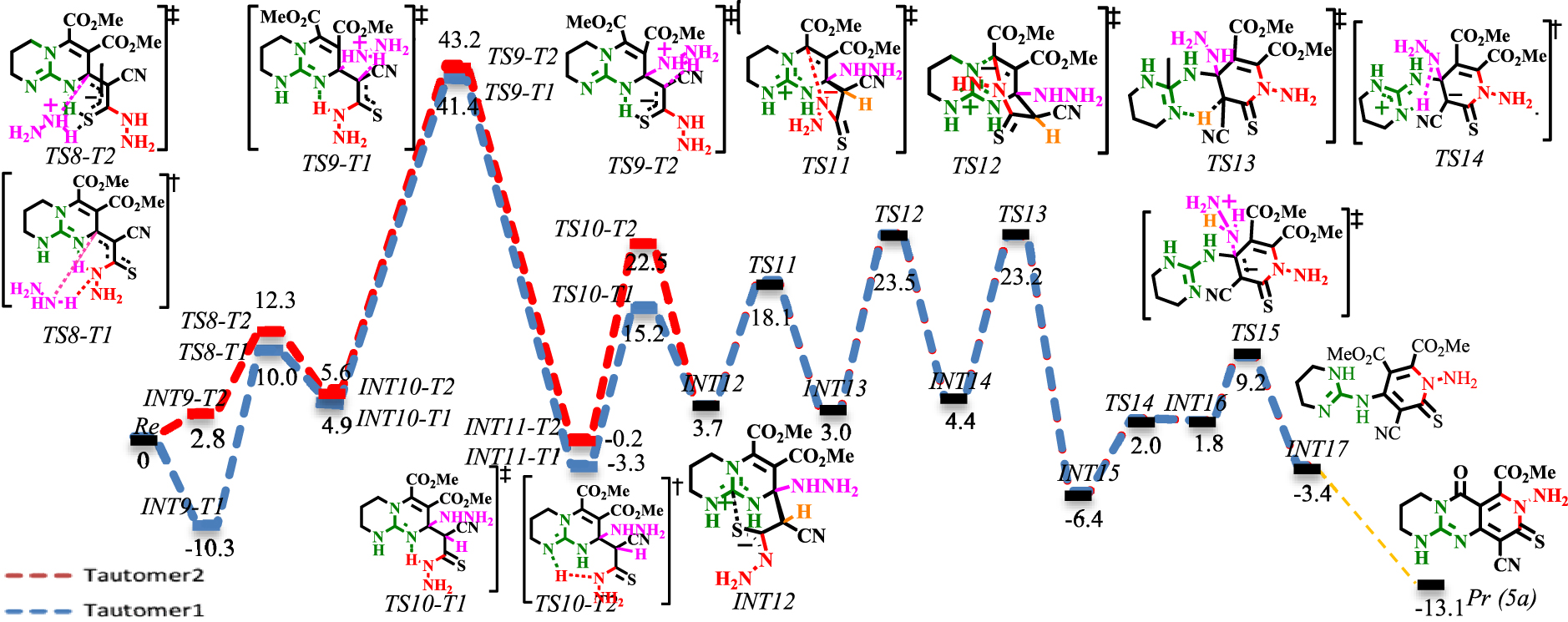
Schéma 7. Profil énergétique de l' addition N- Michael. Les valeurs sont des énergies relatives (𝛥 E298 dans le solvant éthanol, kcal⋅mol−1 ) calculées au niveau M06−2X / 6-311 + G ** // M06−2X / 6-31G *.Schéma 8. Profil énergétique de l' addition de N- Michael sous solvatation explicite de MeOH (lignes vertes et violettes). Les valeurs sont des énergies relatives (𝛥 E298 dans le solvant éthanol, kcal⋅mol−1 ) calculées au niveau M06−2X / 6-311 + G ** // M06−2X / 6-31G *.
3.2.2. Étape d'ajout de N- Michael
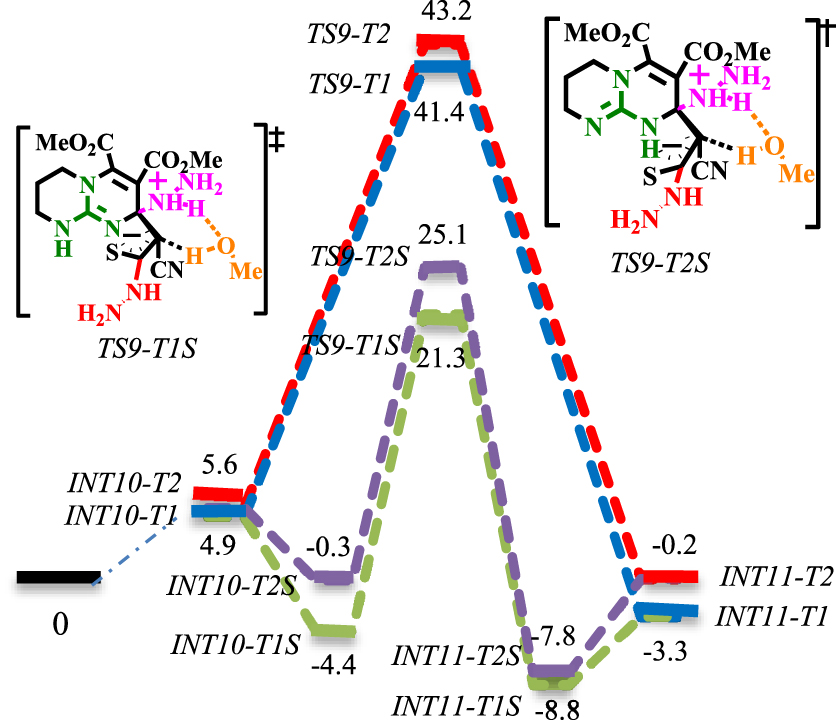
Les profils de réaction pour l' addition de N- Michael sont présentés dans les schémas 7 et 8. Le mécanisme bimoléculaire (lignes rouges et bleues dans les schémas 7 et 8) a été comparé avec le mécanisme termoléculaire impliquant une solvatation explicite de MeOH (lignes vertes et violettes dans le schéma 8).
Schéma 9. Présentation schématique des fonctionnalités CN3 .
En raison du déplacement 1,3-H de l'azote dans le noyau CN3, il est envisagé que le thioamide résultant peut faire avancer la réaction via les tautomères 1 et 2 (pour le mécanisme de tautomérisation, voir le schéma S3 dans les données supplémentaires). Les intermédiaires et états de transition correspondants sont adressés en ajoutant les suffixes T1 et T2 à leurs noms. Comme on le voit dans le schéma 7, le intermoléculaire addition de Michael d'une quantité excessive d'hydrazine (co-catalyseur) à la β carbone insaturé de thiocarbonyle se produit au moyen de [TS8-T1] ‡ avec 20,3 kcal⋅mol-1 barrière. La transformation du proton d'un cation ammonium au carbone of du thiocarbonyle de [TS9-T1] ‡ et [TS9-T2] ‡ rencontre une barrière élevée (36,5 et 37,6 kcal⋅mol-1, respectivement) et ne peut avoir lieu. Comme les états intermédiaires et de transition correspondants sont des complexes ioniques, il a été envisagé que la solvatation explicite par le méthanol puisse résoudre le problème de la barrière élevée (les complexes solvatés par le méthanol seront indiqués en ajoutant un suffixe (S) à leurs noms). Comme le montre le schéma 8, les surfaces d'énergie solvatées MeOH à [TS9-T1S] ‡ et [TS9-T2S] ‡ sont comparativement différentes par rapport à leurs homologues non solvatés ([TS9-T1] ‡ et [TS9-T2] ‡ ). D'autre part, les résultats suggèrent que la transformation du proton via le mécanisme de navette protonique, assistée par MeOH, est significativement plus favorisée que les mécanismes de décalage 1,3-H. La solvatation de [TS9-T1S] ‡ est plus favorable: l'état de transition solvaté [TS9-T1S] ‡ est de 21,3 kcal⋅mol−1et franchit une barrière de 25,43 kcal⋅mol-1 pour donner INT11-T2S. Ensuite, la fonctionnalité de base de Brønsted active le motif nucléophile par abstraction de protons du NH du thioamide. L'énergie d'activation calculée pour l'abstraction de protons à [TS10-T1] ‡ est de 24 kcal⋅mol−1, alors qu'elle est de 30,4 kcal⋅mol−1 à [TS10-T2] ‡. La déprotonation plus facile à [TS10-T1] ‡ est attribuée à la formation d'un état de transition cyclique à six chaînons, qui est moins sollicité que [TS10-T2] ‡. Ensuite, l' addition régiosélective de N- Michael se déroule à [TS11] ‡ avec une barrière de 14,4 kcal⋅mol-1 . Ensuite, la charge négative délocalisée à [TS12] ‡ déclenche la réaction d'ouverture du cycle avec un 20,5 kcal⋅mol−1barrière. Suite à cela, déprotonation basique de Brønsted du proton acide du groupe méthine (via [TS13] ‡), prototropie acide de Brønsted en NH2 NH (via [TS14] ‡) et régénération du co-catalyseur (via [TS15] ‡) délivre INT17. Enfin, INT17 porte une réaction de substitution intramoléculaire entre la fonctionnalité guanidine (base de Lewis) et la fonction ester pour donner 5a (non étudié ici: ligne orange dans le schéma 7 ) [60, 61]. Cette réaction implique l'activité réactive du coeur CN3 .
Le complexe ionique INT12 impliquant l'interaction acide-base de Lewis a un rôle déterminant dans la séquence d'addition de Michael. L'atome de carbone central chargé positivement dans INT12 fonctionne comme un site récepteur d'anion, qui accueille la charge négative délocalisée perpendiculaire au plan du guanidinium. Cette activité mettra les sites réactionnaires en contact étroit, conduisant à l' ajout de N- Michael. Par conséquent, l'exigence d'une quantité excessive du nucléophile en tant que co-catalyseur est rationalisée. Pour démontrer davantage l'interaction de l'acide de Lewis dans INT12, la géométrie optimisée, les orbitales moléculaires et l'analyse NBO sont décrites dans les schémas S4 et S5 (voir les données supplémentaires).
Pour fournir une brève description des activités catalytique-réactif du noyau de triaza CN3, une représentation graphique est donnée dans le schéma 9.
4. Conclusion
En résumé, des calculs DFT ont été utilisés pour acquérir des connaissances sur les activités catalytiques-réactifs de la fonctionnalité guanidine pendant la transformation des guanidines bicycliques en guanidines tricycliques. Il est démontré que la forte acidité de Lewis de l'atome de carbone central dans le noyau CN3 a une influence majeure sur l'avancement de la réaction. Dans l'étape de thioamidation, l'élimination par étapes de MeSH impliquant la complexation acide-base de Lewis avec MeS-et, par la suite, l'abstraction de protons du N − H acide du guanidinium sont énergétiquement plus favorisées que les processus concertés dans lesquels le clivage de la liaison C − SMe est concomitante avec le déplacement 1,3-hydrogène ou la navette protonique assistée par un solvant. Cependant, dans le N-Étape d'addition de Michael, l'acidité de Lewis du guanidinium, en assemblant efficacement à la fois l'azote et le site accepteur de Michael à proximité l'un de l'autre, joue un rôle plus important. Contrairement aux études précédentes, dans lesquelles l'interaction acide de Lewis était incluse dans des transformations asymétriques pour fournir différents résultats stéréochimiques, l'interaction impliquée dans l' étape d'addition N- Michael fournit non seulement une sélectivité, mais déclenche également la réactivité. Sinon, en l'absence d'un tel mode d'activation, le processus doit être arrêté dans l'étape de thioamidation. De plus, des calculs en bon accord avec les résultats expérimentaux ont révélé le rôle central du solvant méthanol ainsi qu'une quantité excessive de N- nucléophiles comme co-catalyseur dans la réalisation de la réaction.
Conflit d'intérêt
Les auteurs ne déclarent aucun conflit d'intérêt.
Données supplémentaires
Des informations complémentaires pour cet article sont disponibles sur le site Web de la revue sous https://doi.org/10.5802/crchim.16 ou auprès de l'auteur. Les détails expérimentaux et les protocoles de randomisation sont fournis.
Bibliographie
[1] D. Leow; C. H. Tan Chiral guanidine catalyzed enantioselective reactions, Chem. Asian J., Volume 4 (2009), pp. 488-507
[2] M. P. Coles Bicyclic-guanidines,-guanidinates and-guanidinium salts: Wide ranging applications from a simple family of molecules, Chem. Commun., Volume 25 (2009), pp. 3659-3676
[3] M. Terada Axially chiral guanidines as efficient Brønsted base catalysts for enantioselective transformations, J. Synth. Org. Chem. Japan, Volume 68 (2010), pp. 1159-1168
[4] T. Ishikawa Guanidine chemistry, Chem. Pharm. Bull., Volume 58 (2010), pp. 1555-1564
[5] X. Fu; C. H. Tan Mechanistic considerations of guanidine-catalyzed reactions, Chem. Commun., Volume 47 (2011), pp. 8210-8222
[6] T. R. Rauws; B. U. Maes Transition metal-catalyzed N-arylations of amidines and guanidines, Chem. Soc. Rev., Volume 41 (2012), pp. 2463-2497
[7] J. E. Taylor; S. D. Bull; J. M. Williams Amidines, isothioureas, and guanidines as nucleophilic catalysts, Chem. Soc. Rev., Volume 41 (2012), pp. 2109-2121
[8] P. Selig Guanidine organocatalysis, Synthesis, Volume 45 (2013), pp. 703-718
[9] X. Fu; C.-H. Tan Mechanistic considerations of guanidine-catalyzed reactions, Chem. Commun., Volume 47 (2011), pp. 8210-8222
[10] R. Salvio The guanidinium unit in the catalysis of phosphoryl transfer reactions: From molecular spacers to nanostructured supports, Chem. Eur. J., Volume 21 (2015), pp. 10960-10971
[11] H. Xue; D. Jiang; H. Jiang; C. W. Kee; H. Hirao; T. Nishimura; M. Wah Wong; C.-H. Tan Mechanistic insights into bicyclic guanidine-catalyzed reactions from microscopic and macroscopic perspectives, J. Org. Chem., Volume 80 (2015), pp. 5745-5752
[12] R. Salvio; C. Alessandro Guanidinium promoted cleavage of phosphoric diesters: Kinetic investigations and calculations provide indications on the operating mechanism, J. Org. Chem., Volume 82 (2017), pp. 10461-10469
[13] X. Fu; Z. Jiang; C. H. Tan Bicyclic guanidine-catalyzed enantioselective phospha-Michael reaction: synthesis of chiral β-aminophosphine oxides and β-aminophosphines, Chem. Commun., Volume 47 (2007), pp. 5058-5060
[14] Z. Jiang; W. Ye; Y. Yang; C. H. Tan Rate acceleration of triethylamine-mediated guanidine-catalyzed enantioselective Michael reaction, Adv. Synth. Catal., Volume 350 (2008), pp. 2345-2351
[15] B. Cho; C. H. Tan; M. W. Wong Sequential catalytic role of bifunctional bicyclic guanidine in asymmetric phosphaMichael reaction, Org. Biomol. Chem., Volume 9 (2011), pp. 4550-4557
[16] B. Cho; C. H. Tan; M. W. Wong Origin of asymmetric induction in bicyclic guanidine-catalyzed thio-Michael reaction: a bifunctional mode of Lewis acid-Brønsted acid activation, J. Org. Chem., Volume 77 (2012), pp. 6553-6562
[17] E. J. Corey; M. J. Grogan Enantioselective synthesis of α-amino nitriles from N-benzhydryl imines and HCN with a chiral bicyclic guanidine as catalyst, Org. Lett., Volume 1 (1999), pp. 157-160
[18] S. Kobayashi; R. Yazaki; K. Seki; Y. Yamashita The fluorenone imines of glycine esters and their phosphonic acid analogues, Angew. Chem. Int. Ed., Volume 47 (2008), pp. 5613-5615
[19] Y. Pan; Y. Zhao; T. Ma; Y. Yang; H. Liu; Z. Jiang; C. H. Tan Enantioselective synthesis of α-fluorinated β-amino acid derivatives by an asymmetric mannich reaction and selective deacylation/decarboxylation reactions, Chem. Eur. J., Volume 16 (2010), pp. 779-782
[20] C. Xie; Y. Dai; H. Mei; J. Han; V. A. Soloshonok; Y. Pan Asymmetric synthesis of quaternary α-fluoro-β-keto-amines via detrifluoroacetylative Mannich reactions, Chem. Commun., Volume 51 (2015), pp. 9149-9152
[21] H. Liu; F. Z. R. de Souza; L. Liu; B. S. Chen The use of marine-derived fungi for preparation of enantiomerically pure alcohols, Appl. Microbiol. Biotechnol., Volume 102 (2018), pp. 1317-1330
[22] P. Hammar; C. Ghobril; C. Antheaume; A. Wagner; R. Baati; F. Himo Theoretical mechanistic study of the TBD-catalyzed intramolecular aldol reaction of ketoaldehydes, J. Org. Chem., Volume 75 (2010), pp. 4728-4736
[23] S. Ding; X. Liu; W. Xiao; M. Li; Y. Pan; J. Hu; N. Zhang 1, 1, 3, 3-Tetramethylguanidine immobilized on graphene oxide: A highly active and selective heterogeneous catalyst for Aldol reaction, Catal. Commun., Volume 92 (2017), pp. 5-9
[24] H. Li; J. Wu; S. Brunel; C. Monnet; R. Baudry; P. Le Perchec Polymerization of lactides and lactones by metal-free initiators, Ind. Eng. Chem. Res., Volume 44 (2005), pp. 8641-8643
[25] R. C. Pratt; B. G. Lohmeijer; D. A. Long; R. M. Waymouth; J. L. Hedrick Triazabicyclodecene: a simple bifunctional organocatalyst for acyl transfer and ring-opening polymerization of cyclic esters, J. Am. Chem. Soc., Volume 128 (2006), p. 4556-4557
[26] B. G. Lohmeijer; R. C. Pratt; F. Leibfarth; J. W. Logan; D. A. Long; A. P. Dove; F. Nederberg; J. Choi; C. Wade; R. M. Waymouth; J. L. Hedrick Guanidine and amidine organocatalysts for ring-opening polymerization of cyclic esters, Macromolecules, Volume 39 (2006), pp. 8574-8583
[27] M. K. Kiesewetter; M. D. Scholten; N. Kirn; R. L. Weber; J. L. Hedrick; R. M. Waymouth Cyclic guanidine organic catalysts: what is magic about triazabicyclodecene?, J. Org. Chem., Volume 74 (2009), pp. 9490-9496
[28] B. A. Chan; S. Xuan; M. Horton; D. Zhang 1, 1, 3, 3-Tetramethylguanidine-promoted ring-opening polymerization of N-butyl N-carboxyanhydride using alcohol initiators, Macromolecules, Volume 49 (2016), pp. 2002-2012
[29] T. Genski; G. Macdonald; X. Wei; N. Lewis; K. R. J Synthesis and application of novel bicyclic guanidines: N-alkylation of 1, 5, 7-triazabicyclo
[4.4. 0] dec-5-ene, Arkivoc, Volume 3 (2000), pp. 266-273
[30] J. C. McManus; J. S. Carey; R. J. Taylor Enantiopure guanidine bases for enantioselective enone epoxidations: 1, acyclic guanidines, Synlett, Volume 2003 (2003), pp. 0365-0368
[31] J. C. McManus; T. Genski; J. S. Carey; R. J. Taylor Enantiopure guanidine bases for enantioselective enone epoxidations: 2, cyclic guanidines, Synlett, Volume 2003 (2003), pp. 369-0371
[32] M. T. Allingham; A. Howard-Jones; P. J. Murphy; D. A. Thomas; P. W. Caulkett Synthesis and applications of C-symmetric guanidine bases, Tetrahedron Lett., Volume 44 (2003), pp. 8677-8680
[33] A. Strecker Untersuchungen über die chemischen beziehungen zwischen guanin, xanthin, theobromin, caffeïn und kreatinin, Justus Liebigs Ann. Chem., Volume 118 (1861), pp. 151-177
[34] G. A. Olah; A. Burrichter; G. Rasul; M. Hachoumy; G. S. Prakash H, C, N NMR and Ab Initio/IGLO/GIAO-MP study of mono-, di-, tri-, and tetraprotonated guanidine1, J. Am. Chem. Soc., Volume 119 (1997), pp. 12929-12933
[35] P. Selig Guanidine organocatalysis, Synthesis, Volume 45 (2013), pp. 703-718
[36] S. E. Denmark; T. W. Wilson N-silyl oxyketene imines are underused yet highly versatile reagents for catalytic asymmetric synthesis, Nat. Chem., Volume 2 (2010), 937 pages
[37] P. Gros; P. Le Perchec; J. P. Senet Reaction of epoxides with chlorocarbonylated compounds catalyzed by hexaalkylguanidinium chloride, J. Org. Chem., Volume 59 (1994), pp. 4925-4930
[38] S. Ullrich; Z. Nazir; A. Büsing; U. Scheffer; D. Wirth; J. W. Bats; G Dürner; M. W. Göbel Cleavage of phosphodiesters and of DNA by a bis (guanidinium) naphthol acting as a metal-free anion receptor, ChemBioChem, Volume 12 (2011), pp. 1223-1229
[39] D. Wirth-Hamdoune; S. Ullrich; U. Scheffer; T. Radanović; G. Dürner; M. W. Göbel A bis (guanidinium) alcohol attached to a hairpin polyamide: Synthesis, DNA binding, and plasmid cleavage, ChemBioChem, Volume 17 (2016), pp. 506-514
[40] F. Foulon; B. Fixari; D. Picq; P. Le Perchec Catalytic decomposition of alkyl chloroformates by hexabutylguanidinium chloride, Tetrahedron Lett., Volume 38 (1997), pp. 3387-3390
[41] C. X. Yan; F. Yang; X. Yang; D. G. Zhou; P. P. Zhou Insights into the diels–alder reaction between 3-vinylindoles and methyleneindolinone without and with the assistance of hydrogen-bonding catalyst bisthiourea: Mechanism, origin of stereoselectivity, and role of catalyst, J. Org. Chem., Volume 82 (2017), pp. 3046-3061
[42] L. Falivene; L. Cavallo Guidelines to select the N-heterocyclic carbene for the organopolymerization of monomers with a polar group, Macromolecules, Volume 50 (2017), pp. 1394-1401
[43] K. Blise; M. W. Cvitkovic; N. J. Gibbs; S. F. Roberts; R. M. Whitaker; G. E. Hofmeister; D. Kohen A theoretical mechanistic study of the asymmetric desymmetrization of a cyclic meso-anhydride by a bifunctional quinine sulfonamide organocatalyst, J. Org. Chem., Volume 82 (2017), pp. 1347-1355
[44] Y. Wang; M. Tang; Y. Wang; D. Wei Insights into stereoselective aminomethylation reaction of α, β-unsaturated aldehyde with N, O-acetal via N-heterocyclic carbene and brønsted acid/base cooperative organocatalysis, J. Org. Chem., Volume 81 (2016), pp. 5370-5380
[45] D. M. Walden; O. M. Ogba; R. C. Johnston; P. H. Y. Cheong Computational insights into the central role of nonbonding interactions in modern covalent organocatalysis, Acc. Chem. Res., Volume 49 (2016), pp. 1279-1291
[46] Q. Peng; R. S. Paton Catalytic control in cyclizations: From computational mechanistic understanding to selectivity prediction, Acc. Chem. Res., Volume 49 (2016), pp. 1042-1051
[47] S. M. Taimoory; T. Dudding An evolving insight into chiral H-bond catalyzed Aza-Henry reactions: A cooperative role for noncovalent attractive interactions unveiled by density functional theory, J. Org. Chem., Volume 81 (2016), pp. 3286-3295
[48] B. Cho; M. Wong Unconventional bifunctional Lewis-Brønsted acid activation mode in bicyclic guanidine-catalyzed conjugate addition reactions, Molecules, Volume 20 (2015), pp. 15108-15121
[49] H. Xue; D. Jiang; H. Jiang; C. W. Kee; H. Hirao; T. Nishimura; M. W. Wong; C. H. Tan Mechanistic insights into bicyclic guanidine-catalyzed reactions from microscopic and macroscopic perspectives, J. Org. Chem., Volume 80 (2015), pp. 5745-5752
[50] A. Alizadeh; A. H. Vahabi; A. Bazgir; H. R. Khavasi; Z. Zhu; L. G. Zhu Determinative role of ring size and substituents in highly selective synthesis of functionalized bicyclic guanidine and tetra substituted thiophene derivatives based on salt adducts afforded by cyclic thioureas and ketene dithioacetal, Tetrahedron, Volume 72 (2016), pp. 1342-1350
[51] M. W. Wong; A. M. E. Ng Asymmetric michael addition using bifunctional bicyclic guanidine organocatalyst: A theoretical perspective, Aust. J. Chem., Volume 67 (2014), pp. 1100-1109
[52] H. Xue; C. H. Tan; M. W. Wong Guanidine-catalyzed asymmetric Strecker reaction: modes of activation and origin of stereoselectivity, Can. J. Chem., Volume 94 (2016), pp. 1099-1108
[53] A. V. Marenich; C. J. Cramer; D. G. Truhlar Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, Volume 113 (2009), pp. 6378-6396
[54] I. Mayer Bond order and valence indices: A personal account, J. Comput. Chem., Volume 28 (2007), pp. 204-221
[55] F. Neese The ORCA program system, Wiley Interdiscip. Rev. Comput. Mol. Sci., Volume 2 (2012), pp. 73-78
[56] S. Cukierman Et tu, Grotthuss and other unfinished stories, Biochim. Biophys. Acta (BBA)-Bioenerg., Volume 1757 (2006), pp. 876-885
[57] N. Susperregui; D. Delcroix; B. Martin-Vaca; D. Bourissou; L. Maron Ring-opening polymerization of ε-caprolactone catalyzed by sulfonic acids: computational evidence for bifunctional activation, J. Org. Chem., Volume 75 (2010), pp. 6581-6587
[58] H. W. Horn; G. O. Jones; D. S. Wei; K. Fukushima; J. M. Lecuyer; D. J. Coady; J. L. Hedrick; J. E. Rice Mechanisms of organocatalytic amidation and trans-esterification of aromatic esters as a model for the depolymerization of poly (ethylene) terephthalate, J. Phys. Chem. A, Volume 116 (2012), pp. 12389-12398
[59] R. C. Pratt; B. G. Lohmeijer; D. A. Long; R. M. Waymouth; J. L. Hedrick Triazabicyclodecene: a simple bifunctional organocatalyst for acyl transfer and ring-opening polymerization of cyclic esters, J. Am. Chem. Soc., Volume 128 (2006), p. 4556-4557
[60] M. K. Kiesewetter; M. D. Scholten; N. Kirn; R. L. Weber; J. L. Hedrick; R. M. Waymouth Cyclic guanidine organic catalysts: what is magic about triazabicyclodecene?, J. Org. Chem., Volume 74 (2009), pp. 9490-9496
[61] S. E. Denmark; G. L. Beutner; T. Wynn; M. D. Eastgate Lewis base activation of Lewis acids: catalytic, enantioselective addition of silyl ketene acetals to aldehydes, J. Am. Chem. Soc., Volume 127 (2005), pp. 3774-3789