Dérivés Δ9,11 d'oestrone comme agents antiprolifératifs potentiels: synthèse, évaluation biologique in vitro et études d'amarrage
Comptes Rendus. Chimie, 2020, 23, no. 2, p. 201-217
Catarina Canário ; Mariana Matias ; Vanessa de Brito ; Adriana O. Santos ; Amílcar Falcão ; Samuel Silvestre ; Gilberto Alves
(traduction post-éditée par N. Bacaër, suggestions d'amélioration : nicolas.bacaer@ird.fr)
Résumé
Une série de dérivés Δ9,11 d'estrone avec des modifications des anneaux A et D ont été synthétisés et évalués en tant qu'agents antiprolifératifs. La cytotoxicité a été évaluée dans six lignées cellulaires (MCF-7, T47-D, LNCaP, HepaRG, Caco-2 et NHDF) par le test de bromure de 3-(4,5-diméthylthiazol-2-yl)-2,5-diphényltétrazolium, et une analyse de la distribution du cycle cellulaire a été réalisée par cytométrie en flux. Certains composés ont présenté une cytotoxicité pertinente, en particulier l'estrone Δ9,11, qui était la plus active contre les cellules HepaRG (IC50=6.67 μM). Outre la pertinence de la double liaison dans le cycle C, la présence d'un groupe 16E-benzylidène a augmenté l'effet antiprolifératif sur les cellules MCF-7 et T47-D. De plus, l'introduction d'iode en positions 2 et 4 de l'estrone semble induire une cytotoxicité sélective pour les cellules HepaRG. Les expériences de cytométrie en flux ont mis en évidence une réduction de 34% de la viabilité des cellules HepaRG après le traitement avec l'estrone Δ9,11 et un arrêt du cycle cellulaire à la phase G0/G1. Une activité œstrogénique a également été observée pour ce composé à 0,1 μM dans les cellules T47-D et les études d'amarrage moléculaire ont estimé une interaction marquée entre ce composé et le récepteur α des œstrogènes .
1. Introduction
L'incidence du cancer a augmenté au fil des ans. On prévoit que 27,5 millions de nouveaux cas de cancer apparaîtront dans le monde chaque année d'ici 2040. Cela représente une augmentation de 61,7% par rapport à 2018 et devrait être plus élevé chez les hommes (augmentation de 67,6%) que chez les femmes (augmentation de 55,3%) [1] . Par conséquent, plusieurs classes de médicaments ont été développées au fil des ans pour traiter le cancer. Par exemple, les taxanes, les anticorps monoclonaux et les stéroïdes sont utilisés en pratique clinique [2, 3, 4].
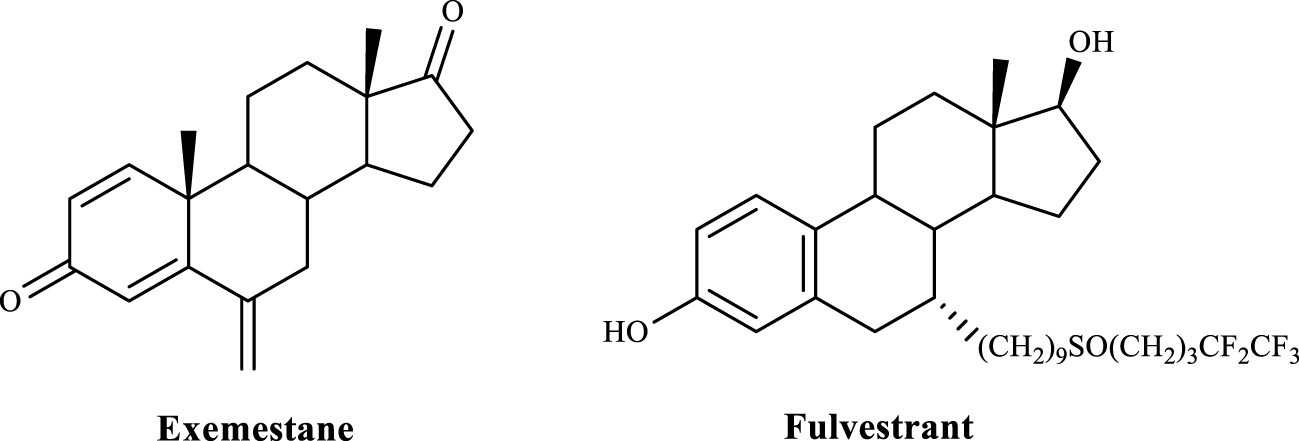
Les hormones stéroïdes sont impliquées dans de nombreuses réponses physiologiques et conditions pathologiques principalement en se liant à leurs récepteurs intracellulaires, à savoir les récepteurs des œstrogènes (RE), qui sont des facteurs de transcription. Par exemple, l'importance des androgènes dans le cancer de la prostate et des œstrogènes dans le cancer du sein a conduit au développement de thérapies bloquant leur action dans ces tumeurs [5]. Dans ce contexte, l'exémestane (un inhibiteur de l'aromatase) [6] et le fulvestrant (un antagoniste sélectif de l'ERα) [7] (figure 1) sont des molécules d'intérêt clinique dans le traitement des cancers du sein hormono-dépendants [8, 9]. Par conséquent, il est essentiel de mettre au point des moyens plus sûrs et plus efficaces de prévenir et de traiter ces cancers hormono-dépendants, compte tenu de l'impact significatif de ces maladies sur la santé humaine et de leur importance économique et sociale [10]. Bien que l'utilisation d'hormones et d'analogues stéroïdiens ait été associée à des cancers hormono-dépendants, des preuves suggèrent également qu'ils peuvent être importants dans le traitement d'autres types de tumeurs telles que les cancers du poumon, du cerveau et du foie [9, 11].
Figure 1.
Stéroïdes pertinents utilisés dans la pratique clinique comme agents anticancéreux.
Compte tenu de l'importance des stéroïdes dans le traitement du cancer, et comme cela a été largement démontré dans la littérature, l'estrone (E1) et le 17ß-estradiol (E2) ont été utilisés comme matières premières pour la conception et le développement de nouveaux anticancéreux plus prometteurs. candidats médicaments avec différentes cibles d'action [8, 12, 13, 14]. Par exemple, la présence d'un 16 α-hydroxyle dans les dérivés d'E1 semble être associée à une cytotoxicité élevée et à une interaction réduite avec ER a [15]. De plus, l’introduction de groupes aryle dans le C-16 de l’échafaudage stéroïdien a conduit à des effets antiprolifératifs plus élevés [16,17]. De plus, la présence de groupes 2-éthyl-3-O-sulfamoyle dans le noyau estrane a permis une amélioration de l'activité antimitotique des analogues E1 [18]. Les dérivés E1 3-O-éther contenant le cycle pipérazine ont également présenté une forte activité cytotoxique contre les lignées cellulaires du cancer de la prostate [19]. Fait intéressant, différents analogues épimères C-13 de la 16ß-(m-carbamoylbenzyl)-E2 ont montré une inhibition de la 17ß-hydroxystéroïde déshydrogénase de type 1 (17ß-HSD1) et un faible effet œstrogénique sur les cellules cancéreuses du sein sensibles aux œstrogènes [20]. Concernant les modifications du cycle C, par exemple, la présence d'une double liaison Δ9,11 combinée à des substitutions 2 et 4 dans le noyau E1 était également pertinente pour le développement d'agents antiprolifératifs prometteurs [21].
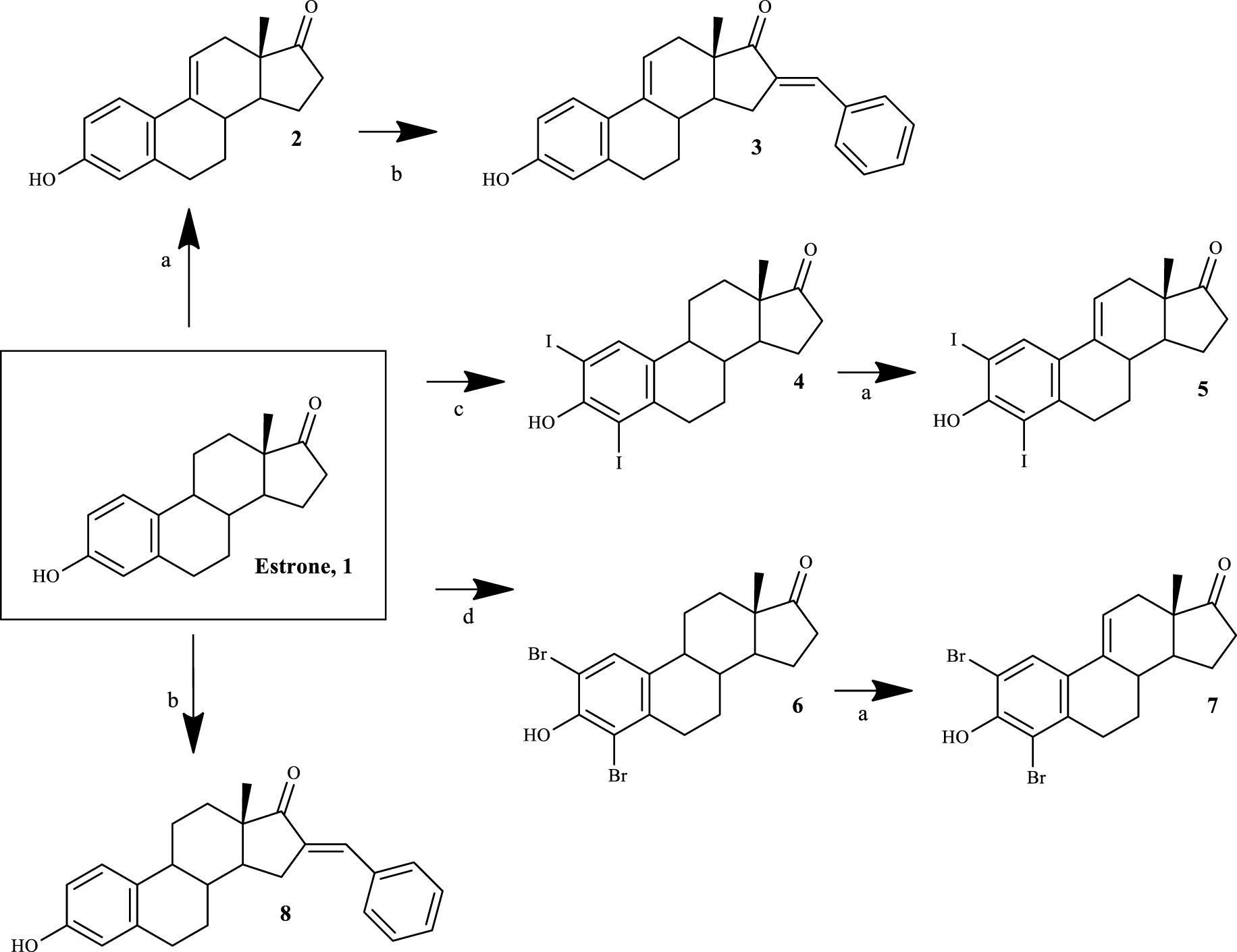
Ces découvertes, en plus de notre intérêt continu pour la chimie stéroïdienne et la bioactivité [8, 22, 23, 24, 25] et la nécessité de développer des agents anticancéreux améliorés, nous ont motivés à préparer et à évaluer in vitro les effets de prolifération cellulaire de dérivés E1. Plus précisément, nous rapportons ici la synthèse chimique de dérivés Δ9,11-E1 avec des modifications A-(2,4-diiodo et 2,4-dibromo) et D (16-benzylidène) et leur évaluation biologique (prolifération cellulaire et essais de viabilité, E dosage-screening et analyse la distribution du cycle cellulaire). Des études d'amarrage sur ERα, la stéroïde sulfatase (ST) et la 17ß-HSD1, qui sont des cibles potentielles pertinentes de ces dérivés de Δ9,11-E1, ont également été réalisées.
2. Expériences
2.1. Chimie
Tous les produits chimiques reçus des fournisseurs ont été utilisés sans autre purification. Les réactifs ont été achetés auprès des fournisseurs suivants: E1: Cayman Chemical (Michigan, USA); méthanol (MeOH): Fisher Chemical (MA, USA); N-bromosuccinimide (NBS): Alfa Aesar (Massachusetts, USA); benzène (PhH): Merck (NJ, USA); benzaldéhyde (BZ): Acros Organics (New Jersey, USA); éthanol (EtOH) 99,9%: Manuel Vieira & Ca (Torres Novas, Portugal). Les réactifs 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ), morpholine, E2, 5-fluorouracile (5-FU) et diméthylsulfoxyde (DMSO) ainsi que les produits chimiques restants mentionnés dans le du texte, comprenant de l'éther de pétrole (PE) 40 à 60°C, ont été obtenus auprès de Sigma-Aldrich (St. Louis, MO, USA). DMSO deutéré (DMSO-d6) et du chloroforme deutéré (CDCl3) ont été achetés auprès Armar Chemicals (Leipzig, Allemagne). Toutes les réactions ont été surveillées par chromatographie sur couche mince (CCM) en utilisant une plaque de gel de silice / aluminium à dos Al 0,20 mm (Macherey-Nagel 60 F254, Duren, Allemagne) et, après élution, les plaques ont été visualisées sous rayonnement ultraviolet (UV) (254 nm) dans une chambre UV CN-15.LC. Un mélange EtOH / acide sulfurique concentré (95: 5, v: v) a été utilisé pour traiter les plaques, suivi d'un chauffage à 120°.L'évaporation des solvants a été réalisée en utilisant un séchoir rotatif sous vide de Büchi (R-215). Les points de fusion (pf) ont été enregistrés sur un appareil de point de fusion Büchi B-540 et ne sont pas corrigés. Les spectres infrarouges (IR) ont été collectés sur un Thermoscientific Nicolet iS10 équipé d'un cristal de réflexion totale atténuée au diamant à température ambiante dans la plage de 4000 à 400 cm-1 en effectuant en moyenne 16 balayages à une résolution spectrale de 2 cm-1 . Spectres de résonance magnétique nucléaire (RMN) (RMN 1H et RMN 13C) ont été acquises sur un spectromètre Bruker Avance 400 MHz et ont été traitées avec le logiciel TOPSPIN 4.07 (Bruker, Fitchburg, WI, USA). Les déplacements chimiques sont rapportés en parties par million (ppm) par rapport au tétraméthylsilane (TMS) ou au solvant comme étalon interne. Les constantes de couplage (valeurs J) sont exprimées en hertz (Hz) et les multiplicités de division sont décrites comme s = singulet, brs = singulet large, d = doublet, dd = doublet double et m = multiplet. La spectrométrie de masse à haute résolution (ESI-HRMS) a été réalisée par le service de microanalyse sur un instrument QSTAR XL (Salamanque, Espagne).
2.1.1. Synthèse de 3-hydroxyestra-1,3,5 (10), 9 (11)-tetraen-17-one (2)
Une solution agitée de 1 (540,8 mg, 2 mmol) dans MeOH (80 ml) a été chauffée à 45°C.DDQ (680,9 mg) a été ajouté en une portion et la solution résultante a été agitée vigoureusement pendant 5 h à 45°C sous atmosphère d'azote (N2). Après achèvement de la réaction (contrôle CCM), MeOH a été évaporé et le résidu a été dilué dans 300 ml de dichlorométhane (CH2Cl2), lavé avec 100 ml de sulfite de sodium aqueux à 10% (Na2SO3), 100 ml de une solution aqueuse saturée d'hydrogénocarbonate de sodium (NaHCO3) et 100 ml d'eau (H2O) et séchée sur sulfate de sodium anhydre (Na 2SO4), filtré et évaporé sous pression réduite pour donner le produit brut, qui a été recristallisé dans MeOH pour donner le composé 2 [26, 27] sous forme de cristaux beiges (223,5 mg, rendement 42%); PF 235,2–237°C (éclairé [28] 243–246°C). IR (νmax, cm-1): 814, 1224, 1453, 1605, 1615, 1715, 2832-2964, 3019, 3255; RMN 1H (400 MHz, DMSO-d6) δ : 0,82 (s, 3H, C18-CH3), 6,05 (m, 1H, C11-H), 6,46 (d, 1H, J = 2,5 Hz, C4-H), 6,55 (dd, 1H, J 1 = 8,6 Hz,J2= 2,5 Hz, C2-H), 7,43 (d, 1H, J = 8,7, C1-H), 9,28 (s large, 1H, 3-OH); RMN 13C (100 MHz, DMSO-d6) δ : 14,21, 22,05, 27,31, 29,20, 33,56, 35,72, 37,69, 45,45, 47,02, 113,82, 114,79, 115,22, 125,03, 125,11, 135,24, 137,19, 156,23, 220,42 .
2.1.2. Synthèse de 3-hydroxy-16-phénylméthylidène-estra-1,3,5 (10), 9 (11)-tetraen-17-one (3)
A une solution du composé 2 (134,2 mg, 0,5 mmol) dans MeOH (3,8 ml), on a ajouté du BZ (76,4 uL) et de l'hydroxyde de potassium (KOH) (192 mg). Le mélange réactionnel a été agité pendant 4,5 h à température ambiante. Après achèvement (contrôle TLC), le mélange réactionnel a été dilué dans 150 ml de CH2Cl2et lavé avec 50 ml de H2O, séché sur Na2SO4anhydre et concentré sous pression réduite pour donner le produit brut, qui était recristallisé dans MeOH pour donner le composé 3 sous forme de cristaux bruns (57,2 mg, rendement 32%); PF 263,1–265,2°C IR (νmax, cm-1): 809, 1285, 1360, 1447, 1496, 1604, 1698, 2829-2958, 3021, 3060, 3324; RMN 1H (400 MHz, DMSO-d6) δ : 0,91 (s, 3H, C18-CH3), 6,09 (m, 1H, C11-H), 6,49 (d, 1H, J = 2,3 Hz, C4-H), 6,56 (dd, 1H, J 1 = 8,7 Hz, J2= 2,4 Hz, C2-H), 7,34 (s large, 1H, H-vinyle), 7,46 (m, 4H, C1-H, H'3, H'4, H'5) ; 7.67 (d, 2H, J = 7.5 Hz, H'2, H'6), 9,30 (s, 1H, 3-OH); RMN 13C (100 MHz, DMSO-d6) δ : 14,89, 27,36, 29,21, 29,47, 33,60, 37,35, 45,03, 45,36, 113,86, 114,83, 115,32, 125,01, 125,11, 128,81, 129,45, 130,41, 132,07, 135,08, 135,41, 136,01, 137,19, 156,28, 208,72. HRMS (ESI-TOF): m / z [M + Na] + calculé pour C 25 H 24 O2: 356,1776; trouvé 356,1771.
2.1.3. Synthèse de 2,4-diiodo-3-hydroxyestra-1,3,5 (10)-trien-17-one (4)
A une solution de E1 1 (270,4 mg, 1 mmol) dans PhH (56 mL) ont été ajoutés 302,8 mg d'iode (I2) et 1536 µL de morpholine. La solution a été agitée à température ambiante pendant 17 h. Après ce temps, 60 ml d'une solution aqueuse de HCl à 5% ont été ajoutés et elle a été concentrée sous pression réduite. Le résultat a été dilué dans 150 ml de CH2Cl 2, lavé avec 50 ml d' une solution aqueuse saturée de NaHCO 3, 50 ml de H2O et on sèche sur Na anhydre 2 SO4et évaporée sous pression réduite. Ensuite, le résidu a été purifié par Chromatographie sur colonne [acétate d'éthyle (EA) / PE, 1: 5] pour obtenir le composé 4 [29] sous forme de solide beige (271 mg, rendement 68%); point de fusion 180,1–183°C (lit [29] 200–202°C). IR (νmax, cm-1): 794, 1011, 1083, 1258, 1450, 1707, 1737, 2858-2961, 3296, 3439; 1H-RMN (400 MHz, CDCl3) δ : 0,88 (s, 3H, C18-CH3), 7,60 (s, 1H, C1-H); RMN 13C (100 MHz, CDCl3) δ : 13,96, 21,74, 26,41, 27,47, 31,61, 36,05, 37,28, 37,59, 44,09, 48,03, 50,35, 78,56, 92,19, 136,07, 136,15, 140,83, 151,70, 220,61.
2.1.4. Synthèse de 2,4-diiodo-3-hydroxyestra-1,3,5 (10), 9 (11)-tetraen-17-one (5)
Une solution agitée de 4 (131,1 mg, 0,25 mmol) dans MeOH (9,8 ml) a été chauffée à 45°C.DDQ (85,1 mg) a été ajoutée en une portion et la solution résultante a été agitée vigoureusement pendant 4 h à 45°C sous Atmosphère N2. Après achèvement (contrôle TLC), MeOH a été évaporé et le résidu a été dilué dans 150 ml d'EA et lavé avec 50 ml de Na2SO3(10%, aqueux), 50 ml de solution aqueuse saturée de NaHCO3et 50 ml de H2O, séché sur Na2SO4anhydre et concentré sous pression réduite. Ce produit a été purifié par chromatographie sur colonne (éluant: EA / PE, 1: 1) pour obtenir le composé5 sous forme de solide brun (72,1 mg, rendement 55%); PF 225,1-227,2°C IR (νmax, cm-1): 794, 1014, 1258, 1447, 1711, 1732, 2920-2961, 3442; 1H-RMN (400 MHz, CDCl3) δ : 0,89 (s, 3H, C18-CH3), 6,12 (m, 1H, C11-H), 7,91 (s, 1H, C1-H); RMN 13C (100 MHz, CDCl3) δ : 14,57, 22,65, 28,35, 34,22, 36,42, 36,81, 37,23, 46,38, 47,94, 79,52, 91,76, 119,27, 131,26, 134,43, 135,31, 140,06, 152,52, 221,24. HRMS (ESI-TOF): m / z [M + Na] + calculé pour C 18 H 18 I2O 2: 519,9396; trouvé 519,9396.
2.1.5. Synthèse de 2,4-dibromo-3-hydroxyestra-1,3,5 (10)-trien-17-one (6)
A une solution de E1 1 (540,7 mg, 2 mmol) dans EtOH (27,0 ml), on a ajouté 1,1 g de NBS. La solution a été agitée à température ambiante pendant 29 h. Après ce temps, le solvant a été évaporé sous pression réduite. Le résidu a été dilué dans 150 ml de CH2Cl 2, lavé avec 50 ml d' une solution aqueuse saturée de NaHCO 3, 50 ml de H2O et on sèche sur Na anhydre 2 SO4et concentrée sous pression réduite. Ensuite, le produit a été recristallisé à partir de MeOH pour donner le composé 6 [30] sous forme de cristaux blancs (353 mg, rendement 41%); PF 228,2–229°C (lit [30] 235–236°C). IR (νmax, cm-1): 899, 1164, 1304, 1462, 1543, 1712, 2869-2936, 3235; 1H-RMN (400 MHz, CDCl3) δ : 0,88 (s, 3H, C18-CH3), 7,38 (s, 1H, C1-H); RMN 13C (100 MHz, CDCl3) δ : 13,95, 21,73, 26,33, 26,69, 31,12, 31,59, 36,03, 37,54, 44,13, 48,00, 50,39, 106,68, 113,42, 128,75, 135,23, 136,66, 147,47, 220,61.
2.1.6. Synthèse de 2,4-dibromo-3-hydroxyestra-1,3,5 (10), 9 (11)-tetraen-17-one (7)
Une solution agitée de 6 (53,5 mg, 0,125 mmol) dans MeOH (4,9 ml) a été chauffée à 45°C.DDQ (42,6 mg) a été ajouté en une portion et la solution résultante a été agitée vigoureusement pendant 5,30 h à 45°C sous Atmosphère N2. Après achèvement (contrôle TLC), MeOH a été évaporé puis le résidu a été dilué dans 150 mL d'EA, 50 mL de solution aqueuse de Na2SO3(10%), 50 mL de solution aqueuse saturée de NaHCO3et 50 mL de H2O puis séché sur Na2SO4anhydre et concentré sous pression réduite pour obtenir le composé 7sous forme d'un solide beige (38 mg, 71% de rendement); PF 200,4-202,9°C IR (νmax, cm-1): 796, 1011, 1064, 1260, 1463, 1540, 1717, 2836-2960, 3286; 1H-RMN (400 MHz, CDCl3) δ : 0,89 (s, 3H, C18-CH3), 6,13 (m, 1H, C11-H), 7,69 (s, 1H, C1-H); RMN 13C (100 MHz, CDCl3) δ : 14,59, 22,65, 27,60, 30,70, 34,19, 36,41, 37,25, 46,35, 47,95, 107,59, 113,14, 119,49, 127,81, 130,32, 134,47, 135,98, 148,30, 221,27. HRMS (ESI-TOF): m / z [M + H] + calculé pour C 18 H 18 Br2O 2: 423,9674; trouvé 423,9644.
2.1.7. Synthèse de la 3-hydroxy-16-phénylméthylidène-estra-1,3,5 (10)-tetraen-17-one (8)
A une solution de 1 (135,2 mg, 0,5 mmol) dans MeOH (3,8 ml) ont été ajoutés du BZ (76,4 uL) et du KOH (192 mg). Le mélange a été agité à température ambiante pendant 4 h. Après évaporation de MeOH, le mélange réactionnel a été dilué dans 150 ml de CH2Cl2et lavé avec 50 ml de H2O, séché sur Na2SO4anhydre et concentré sous pression réduite pour donner le produit brut, qui a été recristallisé à partir de MeOH. pour donner le composé 8 [31, 32] sous forme de cristaux blancs (162 mg, rendement 90%); PF 247,5–249,7°C (éclairé [32] 248–250°C). IR (νmax / cm-1): 790, 1276, 1373, 1445, 1612, 1699, 2858-2920, 3019, 3053, 3350; RMN 1H (400 MHz, CDCI3) δ : 1,00 (s, 3H, C18-CH3), 4,82 (s large, 1H, 3-OH), 6,60 (s large, 1H, C4-H), 6,66 (d, 1H, J = 9,1 Hz, C2-H), 7,17 (d, 1H, J = 9,1 Hz, C1-H), 7,39 (m, 3H, H'3, H'4, H'5); 7.48 (brs, 1H, H-vinyl), 7.57 (d, 2H, J = 8.3 Hz, H'2, H'6); RMN 13C (100 MHz, CDCl3) δ : 14,78, 26,19, 27,01, 29,37, 29,68, 31,90, 38,20, 44,26, 48,08, 48,81, 113,10, 115,53, 126,71, 128,91, 129,49, 130,56, 132,33, 133,56, 135,82, 136,20, 138,19, 153,77, 210,04.
2.2. Tests de bioactivité
2.2.1. Culture de cellules
Des lignées de cellules mammaires humaines (MCF-7, T47-D), prostatiques (LNCaP), colon (Caco-2) et fibroblastes [fibroblastes dermiques humains normaux (NHDF)] ont été obtenues auprès de l'American Type Culture Collection (ATCC; Manassas, VA, USA) et hépatiques (HepaRG) ont été acquises auprès de Life Technologies — Invitrogen ™ (via Alfagene, Portugal). Ils ont été cultivés dans des flacons de culture de 75 cm 2 à 37°C dans un incubateur à air humidifié avec 5% de CO2. Milieu Eagle modifié de Dulbecco à haute teneur en glucose (DMEM) supplémenté avec 10% de sérum bovin fœtal (FBS; Sigma-Aldrich, St Louis, MO, USA) et 1% d'antibiotique / antimycotique (10000 unités / ml de pénicilline G, 100 mg / ml de streptomycine et 25 µg / ml d'amphotéricine B) (Ab; Sigma-Aldrich, St Louis, MO, USA) a été utilisé pour cultiver des cellules MCF-7. Pour les cellules Caco-2, DMEM à haute teneur en glucose supplémenté avec 10% de FBS et 1% du mélange antibiotique de 10000 unités / ml de pénicilline G et 100 mg / ml de streptomycine (sp; Sigma-Aldrich, St Louis, MO, USA) a été utilisé. Les cellules LNCaP et T47-D ont été cultivées dans du milieu RPMI 1640 avec 10% de FBS et 1% de sp. Les fibroblastes se sont développés dans un milieu RPMI 1640 supplémenté avec 10% de FBS, 2 mM de L-glutamine, 10 mM de HEPES, 1 mM de pyruvate de sodium et 1% d'Ab. Enfin, les cellules HepaRG ont été ensemencées dans du milieu E de Williams supplémenté avec 10% de FBS, 1% de sp, 5 µg / mL d'insuline et 5 x 10-5 M d'hémisuccinate d'hydrocortisone (Sigma-Aldrich, St Louis, MO, USA).
2.2.2. Préparation de solutions composées
Des solutions mères de composés ont été préparées dans du DMSO à 10 mM et stockées à 4–8°C. La concentration maximale de DMSO dans les études cellulaires était de 1% et les expériences précédentes ont révélé que ce niveau de solvant n'a pas d'effets significatifs sur la prolifération cellulaire (données non présentées) .
2.2.3. Dosages antiprolifératifs
Cytotoxicité des composés 1-8 a été évaluée par la 3-(4,5-diméthylthiazol-2-yl)-2,5-diphényltétrazolium (MTT, Sigma-Aldrich, St Louis, MO, USA) Essai contre MCF-7, Cellules T47-D, LNCaP, HepaRG, Caco-2 et NHDF. Après avoir atteint la confluence proche, les cellules ont été trypsinisées et comptées avec un hémocytomètre au moyen de l'exclusion au bleu trypan des cellules mortes. Ensuite, 100 µL de suspension cellulaire (2 x 104 cellules / ml) ont été ensemencés dans des plaques de culture à 96 puits et laissés à adhérer pendant 48 h. Après adhérence, le milieu a été remplacé par plusieurs solutions des composés de l'étude (30 µM pour les tests de criblage et 0,1, 1, 10, 25, 50 et 100 µM pour les études concentration-réponse) dans le milieu de culture approprié pendant environ 72 h. Après cette période, les cellules ont été lavées avec 100 µL de tampon phosphate salin (PBS; NaCl 137 mM, KCl 2,7 mM, Na2HPO410 mM et KH2PO41,8 mM, pH 7,4), puis 100 µL de la solution MTT (5 mg / ml), préparée dans le milieu sans sérum approprié, a été ajoutée à chaque puits, suivie d'une incubation pendant environ 4 h à 37°Ensuite, le milieu contenant du MTT a été éliminé et les cristaux de formazan ont été dissous dans du DMSO. L'absorbance a été mesurée à 570 nm en utilisant un spectrophotomètre Bio-rad xMark de lecteur de microplaques. Après soustraction de fond, les valeurs de prolifération cellulaire ont été exprimées en pourcentage par rapport à l'absorbance déterminée dans les cellules témoins négatives. Des cellules non traitées ont été utilisées comme contrôle négatif et le médicament clinique 5-FU a été utilisé comme contrôle positif. Chaque expérience a été réalisée en quadruple et répétée indépendamment.
2.2.4. Test de dépistage électronique
Les cellules T47-D (2 x 104 cellules / ml) ont été ensemencées dans des plaques de culture à 96 puits à 100 µM de milieu RPMI 1640 additionné de 10% de FBS et on les laisse attacher. Après une nuit d'incubation, le milieu a été remplacé tous les 3 jours par du milieu RPMI 1640 sans rouge phénol frais supplémenté avec 5% de sérum de veau foetal traité au charbon de bois revêtu de dextrane (DCC-FCS) et contenant les composés à l'étude. Après 6 jours d'exposition, la prolifération des cellules T47-D a été estimée par le test MTT décrit dans la section précédente. 0,1, 0,01 et 0,001 µM étaient les concentrations testées pour E2 et pour les composés sélectionnés synthétisés. Chaque expérience a été réalisée en quadruple et répétée indépendamment. Après soustraction de fond, les valeurs de prolifération cellulaire ont été exprimées en pourcentage par rapport à l'absorbance déterminée dans les cellules témoins négatives.
2.2.5. Analyse cytométrique en flux de la viabilité cellulaire
L'analyse de la viabilité cellulaire sur cellules HepaRG a été réalisée par cytométrie de flux après coloration des cellules mortes à l'iodure de propidium (PI) (solution de PI 1 mg / ml dans 0,1% d'azide de sodium et d'eau; Sigma-Aldrich, St Louis, MO, USA). En bref, 3 ml de suspension de cellules a été ensemencée dans des plaques à 6 puits (5 x 104 cellules / ml) dans un milieu de culture complet. Après 48 h, ils ont été traités avec 50 µM de composé 2. Les cellules non traitées ont été utilisées comme contrôle négatif et le 5-FU a été utilisé comme contrôle positif. Chaque expérience a été réalisée en double et répétée indépendamment. Au bout de 24 h d'incubation, le surnageant de chaque puits a été collecté; les cellules ont été récoltées par trypsinisation et rassemblées avec les surnageants. La suspension cellulaire résultante a été conservée sur de la glace, culottée par centrifugation et remise en suspension dans 400 µL de milieu complet. Ensuite, 395 µL de la suspension cellulaire ont été transférés dans un tube FACS et 5 µL de PI avec EDTA (0,5 µL à 0,123 M) a été ajouté. Un minimum de 20000 événements a été acquis en utilisant un cytomètre de flux BD Accuri C6 (San Jose, USA) dans les canaux de diffusion directe (FSC), de diffusion latérale (SSC) et de fluorescence canal-3 (FL3, pour PI). L'acquisition et l'analyse ont été effectuées avec le logiciel BD Accuri. Dans le tracé de contour FSC / FL3, trois régions ont été créées, une correspondant aux cellules viables (R1), une autre aux cellules mortes (R2) et une troisième à une population cellulaire indéterminée entre les deux autres régions (R3) à l'exclusion des débris qui n'étaient pas considéré dans l'analyse (données non présentées). Le pourcentage de viabilité est le pourcentage de cellules dans R1 par rapport au nombre total d'événements dans R1, R2 et R3.
2.2.6. Analyse cytométrique en flux du cycle cellulaire
Après 24 h de traitement avec 50 µM de composé 2 (plaques 6 puits, 5 × 104 cellules / mL), les cellules HepaRG ont été collectées et lavées avec du PBS et remises en suspension dans 450 µL d'une solution froide de sérum bovin à 0,5% albumine (BSA; Amresco, USA) en PBS avec EDTA (204 µM dans 25 mL), suivie d'une fixation avec 70% d'EtOH et d'une incubation à-20°C. Après au moins 2 jours à-20°C, les cellules fixées ont été lavées deux fois avec du PBS et remises en suspension dans une solution de PI (50 µg / mL) préparée dans 0,5% de BSA dans du PBS avec EDTA puis incubées avec de la ribonucléase A de pancréas bovin à une concentration finale de 0,5 µg / µL (solution dans 50% de glycérol, Tris-HCl 10 mM, pH 8; Sigma-Aldrich, St Louis, MO, USA) pendant 15 min dans l'obscurité. A titre de comparaison, des cellules non traitées ont été utilisées comme contrôle négatif et des cellules traitées avec du 5-FU à 50 uM ont été utilisées comme contrôle positif. Chaque expérience a été réalisée en double et répétée indépendamment. Un minimum de 10000 événements a été acquis à l'aide du logiciel BD Accuri et l'analyse a été effectuée par le logiciel Modfit (Becton Dickinson, San Jose, CA, USA).
2.2.7. Dosage de l'ester succinimidylique de carboxyfluorescéine par cytométrie en flux
Les cellules HepaRG ont été trypsinisées, comptées et ensemencées dans deux plaques de culture à 12 puits (1 ml / puits, 8 × 104 cellules / ml) et on laisse adhérer pendant 48 h. Après cette période, les cellules ont été rincées deux fois avec du PBS puis de l'ester succinimidylique de carboxyfluorescéine (CFSE; BD Horizon, San Jose, USA) a été ajouté à 10 µM et incubé pendant 15 ou 30 min. Après incubation, les puits ont été rincés avec du PBS et le milieu avec le composé 2 (50 µM) a été ajouté, suivi d'une incubation pendant environ 48 et 72 h. Des cellules non traitées ont été utilisées comme contrôle négatif dans chaque plaque. Pendant 15 min d'incubation CFSE, chaque expérience a été réalisée en double. Pendant 30 min d'incubation, une expérience a été réalisée pendant 48 h et une autre pendant 72 h. A la fin de la période d'incubation, les cellules ont été trypsinisées, centrifugées et remises en suspension dans 300 µL de milieu avec 5 µL d'EDTA. Un minimum de 20000 événements a été acquis en utilisant un cytomètre de flux BD Accuri C6 dans les canaux de diffusion vers l'avant (FSC), de diffusion latérale (SSC) et de fluorescence channel-1 (FL1, pour CFSE). L'acquisition et l'analyse ont été effectuées avec le logiciel BD Accuri.
2.2.8. analyses statistiques
Les données ont été exprimées en moyenne ± écart-type (ET). La comparaison entre les groupes a été réalisée en utilisant le test t-étudiant (deux groupes) et l'ANOVA à un facteur (trois groupes) suivis des tests post hoc de Bonferroni pour déterminer les différences statistiquement significatives entre les moyennes. La différence entre les groupes a été considérée comme statistiquement significative pour une valeur p inférieure à 0,05 (p <0,05). La détermination de la CI 50 a été réalisée par analyse d'ajustement sigmoïdal en considérant un niveau de confiance de 95%.
2.3. Études d'amarrage moléculaire
2.3.1. Préparation de protéines pour l'ancrage moléculaire
Les structures cristallines de ERα, ST et 17ß-HSD1 ont été obtenues à partir de la Protein Data Bank (code PDB: 1A52, 1P49 et 3KLM, respectivement) [33, 34, 35]. Les coordonnées de tous les résidus non standard ont été supprimées à l'aide du logiciel Chimera (v. 1.10.1). Ensuite, des hydrogènes non polaires ont été fusionnés dans AutoDockTools (v. 1.5.6) et des charges partielles de Kollman et Gasteiger ont été ajoutées. Enfin, la structure préparée a été convertie du format PDB en PDBQT pour une utilisation postérieure dans l'étude d'amarrage.
2.3.2. Préparation des ligands
Tous les ligands ont été construits en utilisant le logiciel Chem3D (v. 12.0). La minimisation de l'énergie et l'optimisation de la géométrie ont été effectuées par le même logiciel et les structures finales ont été enregistrées sous un format de fichier PDB. Le processus de minimisation de l'énergie a été appliqué dans une plage allant de-20 à-40 kcal·mol-1 . Ensuite, les ligands ont été complètement préparés en choisissant des torsions et les structures ont été converties du format PDB au format PDBQT dans le logiciel AutoDockTools.
2.3.3. Paramètres de la grille
Les paramètres de la grille ont été calculés à l'aide d'AutoDock Vina et d'AutoDockTools en fonction des coordonnées du ligand cristallisé pour chaque cas: E2, N-acétyl-D-glucosamine et 5 a-dihydrotestostérone (DHT), avec la macromolécule respective. La grille était centrée sur le ligand avec les coordonnées suivantes: pour ERα, les coordonnées étaient x = 107,27, y = 13,94, z = 96,38; pour ST, x = 62,033, y =-12,215, z = 52,512; et pour 17ß-HSD1, x = 11,643, y = 9,297, z=-11,887. La taille de la grille était de 20 x 20 x 20 avec un espacement de 1,0 Å.
2.3.4. Simulations d'amarrage
Après la préparation des ligands et des protéines, l'ancrage moléculaire a été effectué par l'exécutable AutoDock Vina, qui utilise un optimiseur global de recherche locale itéré. L'exhaustivité des paramètres des expériences réalisées a été définie comme 8 (par défaut). Les résultats de l'amarrage moléculaire ont été visualisés dans le programme Discovery Studio Visualizer de BIOVIA et dans le logiciel PyMOL.
2.3.5. Validation des performances d'amarrage moléculaire
Les fonctions de notation sont essentielles pour les performances d'amarrage moléculaire. Afin de vérifier ces fonctions, il est nécessaire de valider les performances d'amarrage d'AutoDock Vina. Cette étape est nécessaire pour vérifier les performances par l'analyse de la différence entre les conformations réelles et les mieux notées. Pour que le processus d'ancrage soit considéré comme réussi, la valeur de la distance moyenne quadratique (RMSD) entre ces deux conformations doit être inférieure à 2,0 Å. Dans ce cas, la méthode de validation a été réalisée en re-docking ERα avec E2, ST avec N-acétyl-D-glucosamine et 17ß-HSD1 avec DHT. Des valeurs RMSD faibles ont été obtenues pour tous les cas, ce qui signifie que le processus d'amarrage était fiable et validé.
3. Résultats et discussion
3.1. Chimie
Quatre dérivés Δ9,11-E1 ont été synthétisés par la procédure de synthèse générale décrite dans le schéma 1 . A notre connaissance, trois de ces dérivés ont été synthétisés pour la première fois (composés 3, 5 et 7). Tous les composés ont été caractérisés par analyse spectrale (IR, RMN 1H et 13 C) et un HRMS a également été obtenu pour les nouveaux stéroïdes préparés. Toutes les données spectrales sont en accord avec les structures présentées. Par exemple, la présence d'une double liaison Δ9,11 a été observée à partir du signal du proton C-11 apparaissant entre 6,05 et 6,13 ppm dans le 1Spectres de RMN H, conformément aux résultats décrits dans la littérature [27].
Schéma 1.
Voie synthétique pour préparer des dérivés de Δ9,11-estrone. Réactifs et conditions: (a) DDQ, MeOH, reflux; (b) benzaldéhyde, KOH, MeOH, température ambiante; (c) I 2, morpholine, PhH, température ambiante; (d) NBS, EtOH, température ambiante.
La synthèse des dérivés Δ9,11-E1 peut être réalisée en utilisant l'ion adamantyl carbonium comme agent déshydrogénant [36]. De plus, Brown et al. [37] ont décrit une voie plus simple utilisant la DDQ pour obtenir ces composés à partir de E1 avec un rendement élevé. Plus tard, cette dernière procédure a été améliorée par d'autres groupes de recherche [27]. Bien que la préparation de dérivés Δ9,11-estranaires soit connue depuis les années 1960, les activités biologiques de ce groupe de composés, en particulier leur activité anticancéreuse potentielle, restent relativement inexplorées. Dans ce contexte, Milic et al. [21] ont décrit des résultats prometteurs de cytotoxicité de dérivés Δ9,11-E1 2-et 4-substitués dans différentes lignées cellulaires cancéreuses, témoignant de l'intérêt pour cette modification du cycle C de E1. Sur la base de ces informations, afin d'obtenir des composés aux effets cytotoxiques prometteurs, des modifications du cycle A (positions 2 et 4) de E1 ont été combinées avec la double liaison Δ9,11 . De plus, du fait que la présence d'un groupe 16-arylidène dans le squelette stéroïde est également associée à des propriétés cytotoxiques notables dans plusieurs lignées cellulaires [16, 35], cette modification a également été explorée par nous.
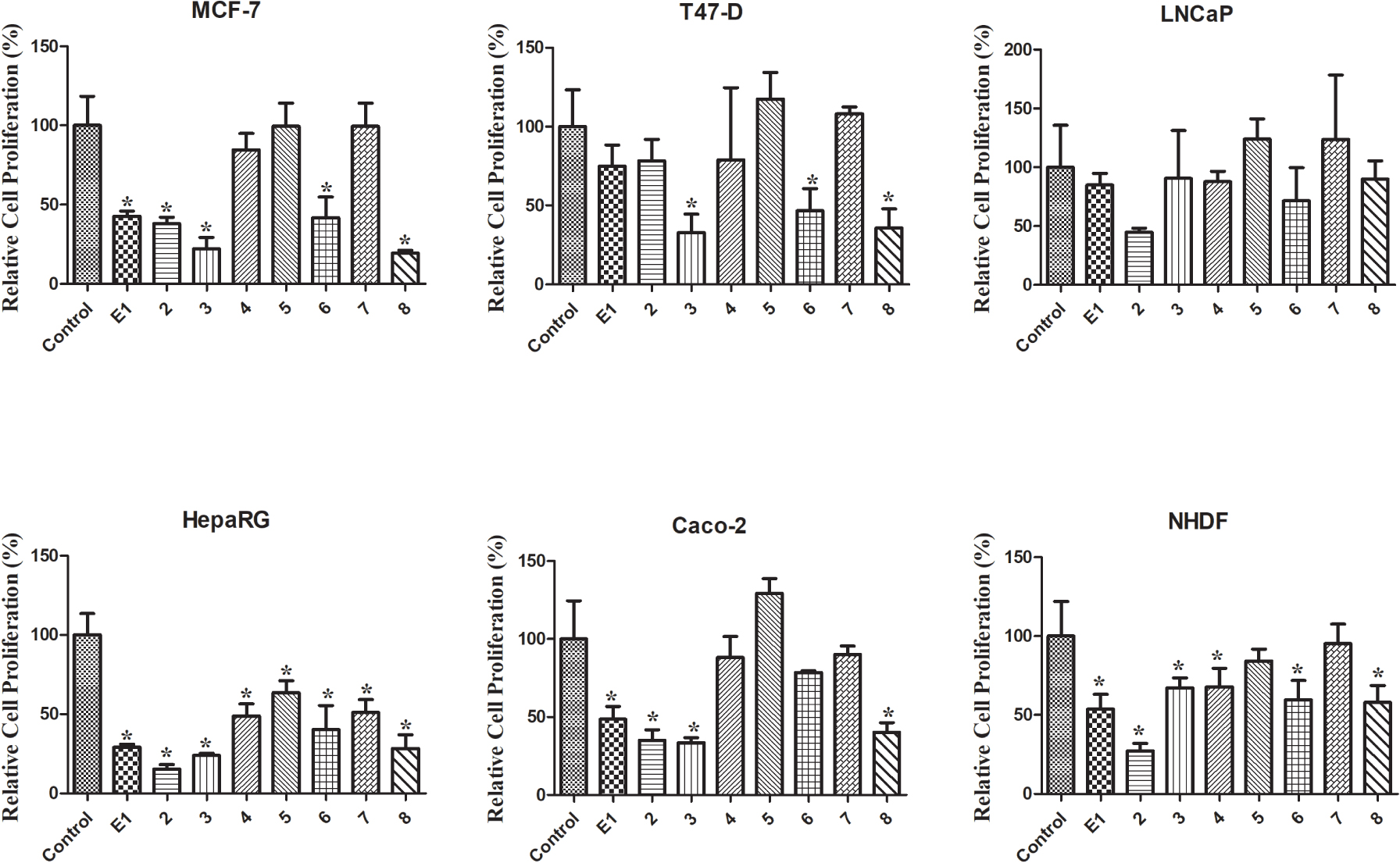
Figure 2.
Prolifération cellulaire relative des cellules MCF-7, T47-D, LNCaP, HepaRG, Caco-2 et NHDF incubées avec les composés synthétisés, pendant 72 h à une concentration de 30 µM, déterminée par le test MTT, quantification spectrophotométrique du formazan à 570 nm . Les données sont exprimées en pourcentage de prolifération cellulaire par rapport au témoin négatif et sont indiquées comme des moyennes ± écart-type et sont représentatives d'au moins deux expériences indépendantes. * p <0,05 vs contrôle.
Ainsi, l'introduction de la double liaison Δ9,11 dans E1 a donné le composé 2 . Ce processus a été réalisé avec succès à l'aide de DDQ, comme décrit dans la littérature [27]. Ensuite, en utilisant BZ et KOH, le composé 3 a été facilement synthétisé par une réaction d'aldol médiée par une base [38], où le stéroïde 16E-benzylidène correspondant a été obtenu [25]. Dans ce contexte, le signal du proton ponté par la méthine en C-16 est apparu à 7,34 ppm dans les spectres RMN 1H [39], et une configuration E a été attribuée à cette double liaison sur la base des rapports précédents [25]. L'iodation du cycle A E1 (en utilisant I2) et la bromation (en utilisant le NBS) ont été effectuées pour obtenir respectivement la 2,4-diiodoestrone (composé 4) [40] et la 2,4-dibromoestrone (composé 6) [30]. Parmi ces deux types d'halogénation aromatique, la bromation était plus simple à réaliser que l'iodation. En fait, deux autres stratégies plus vertes ont été essayées avant l'utilisation réussie de I2/ morpholine / PhH pour l'iodation [40]. Cette dernière procédure était préférable à la combinaison d'iodure de sodium et de chlorite de sodium [41], qui ne permettait la synthèse de la 2-iodoestrone qu'avec de faibles rendements. De plus, en utilisant I 2et le chlorure de cuivre (II) dihydraté (CuCl2.2H2O) [42], après la réaction, il était très difficile de séparer les isomères 2-et 4-iodoestrone par chromatographie sur colonne. Cette iodation nécessite également une atmosphère non oxydante, ce qui prend plus de temps. Ensuite, les intermédiaires 4 et 6 ont été utilisés pour préparer les deux nouveaux dérivés Δ9,11 5 et 7 par DDQ, comme décrit ci-dessus. Fait intéressant, dans des conditions de réaction similaires, un rendement plus élevé du produit a été observé dans la déshydrogénation de la 2,4-dibromoestrone. Enfin, composé 8a également été préparé à partir de E1 par condensation aldolique, visant à améliorer les données de relation structure-relation en comparant sa bioactivité à celle observée pour les composés 3 et même E1.
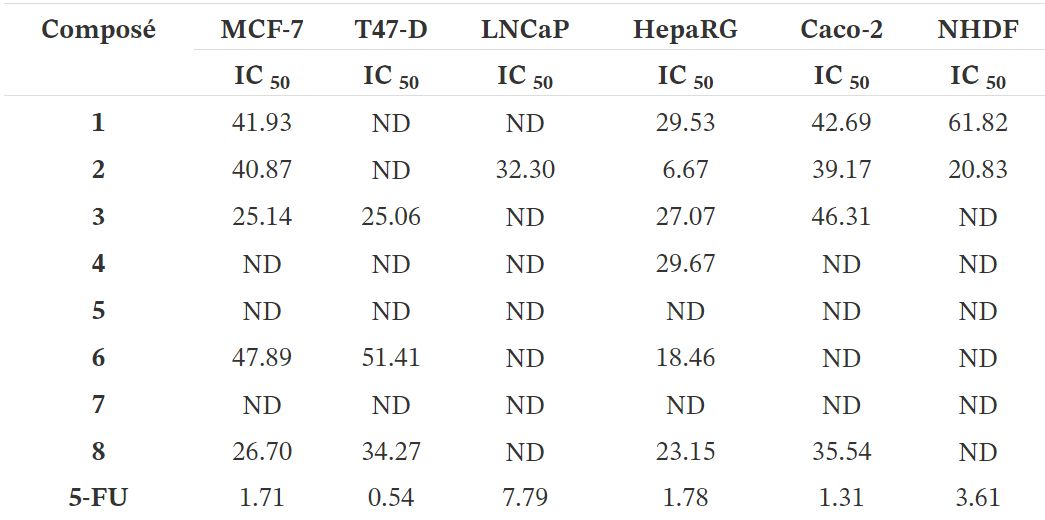
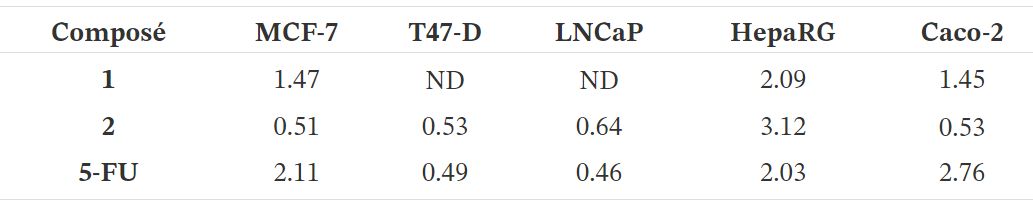
Tableau 1.
Cytotoxicité (IC 50 en µM) des composés synthétisés (1–8) ainsi que du 5-FU contre le sein (MCF-7 et T47-D), prostatique (LNCaP), hépatique (HepaRG) et colon (Caco-2) lignées cellulaires cancéreuses et fibroblastes dermiques humains normaux (NHDF) a
a Les cellules ont été traitées avec différentes concentrations (0,1, 1, 10, 25, 50 et 100 µM) pendant 72 h. Les effets de prolifération cellulaire ont été déterminés par le test MTT. Les données présentées sont représentatives d'au moins deux expériences indépendantes. ND: non déterminé.
3.2. Tests biologiques
3.2.1. Effet de croissance cellulaire
Le test colorimétrique MTT a été effectué pour évaluer la cytotoxicité des composés 1-8 sur hormono-dépendant (MCF-7, T47-D et LNCaP) et hormone-indépendant (HepaRG et Caco-2) les cellules cancéreuses et sur NHDF. Tout d'abord, un criblage à 30 µM a été effectué pour tous les composés dans toutes les lignées cellulaires (figure 2). Lorsque la réduction de la prolifération des cellules était supérieure à 50%, le CI 50 a été déterminée. Comme le montre le tableau 1, on a observé la réduction la plus pertinente de la prolifération cellulaire avec le composé 2 dans les cellules HepaRG (IC 50 = 6,67 µM). Fait intéressant, l'introduction du Δ9,11la double liaison dans E1 a augmenté les effets cytotoxiques pour toutes les lignées cellulaires étudiées, à l'exception des cellules T47-D. La présence du groupe 16E-benzylidène (composé 3) a augmenté les effets cytotoxiques sur les cellules MCF-7 (IC 50 = 25,14 µM) et T47-D (IC 50 = 25,06 µM) par rapport au composé 2 . En comparant la bioactivité des composés 3 et 8, il était intéressant de noter que la présence de la double liaison Δ9,11 dans ces 16E-benzylidènes a également conduit à une augmentation de la cytotoxicité dans les lignées de cellules mammaires mais pas dans les cellules HepaRG et Caco-2. En outre, l'introduction de l' iode dans les positions 2 et 4 de E1 (composé 4) autorisés cytotoxicité sélective contre les cellules HepaRG (IC 50 = 29,67 µM). Le stéroïde dibromé 6 avait généralement une cytotoxicité plus élevée que l'analogue iodé correspondant 4 . En revanche, aucune réduction prononcée de la prolifération cellulaire n'a été observée pour les composés 5 et 7 dans toutes les lignées cellulaires testées. Par conséquent, contrairement à ce qui a été observé pour E1 et composé 2, la présence d'iode et de brome en positions 2 et 4 de Δ9,11-E1 n'était pas un changement structurel favorable pour le développement d'agents antiprolifératifs potentiels.
En ce qui concerne les valeurs de l'indice de sélectivité (SI) (tableau 2), on sait qu'une valeur de 2 ou plus indique une sélectivité élevée pour les cellules cancéreuses [43]. Selon ces informations, la sélectivité du composé 2 vis-à-vis de la lignée cellulaire HepaRG est très intéressante (SI> 3).
Figure 3.
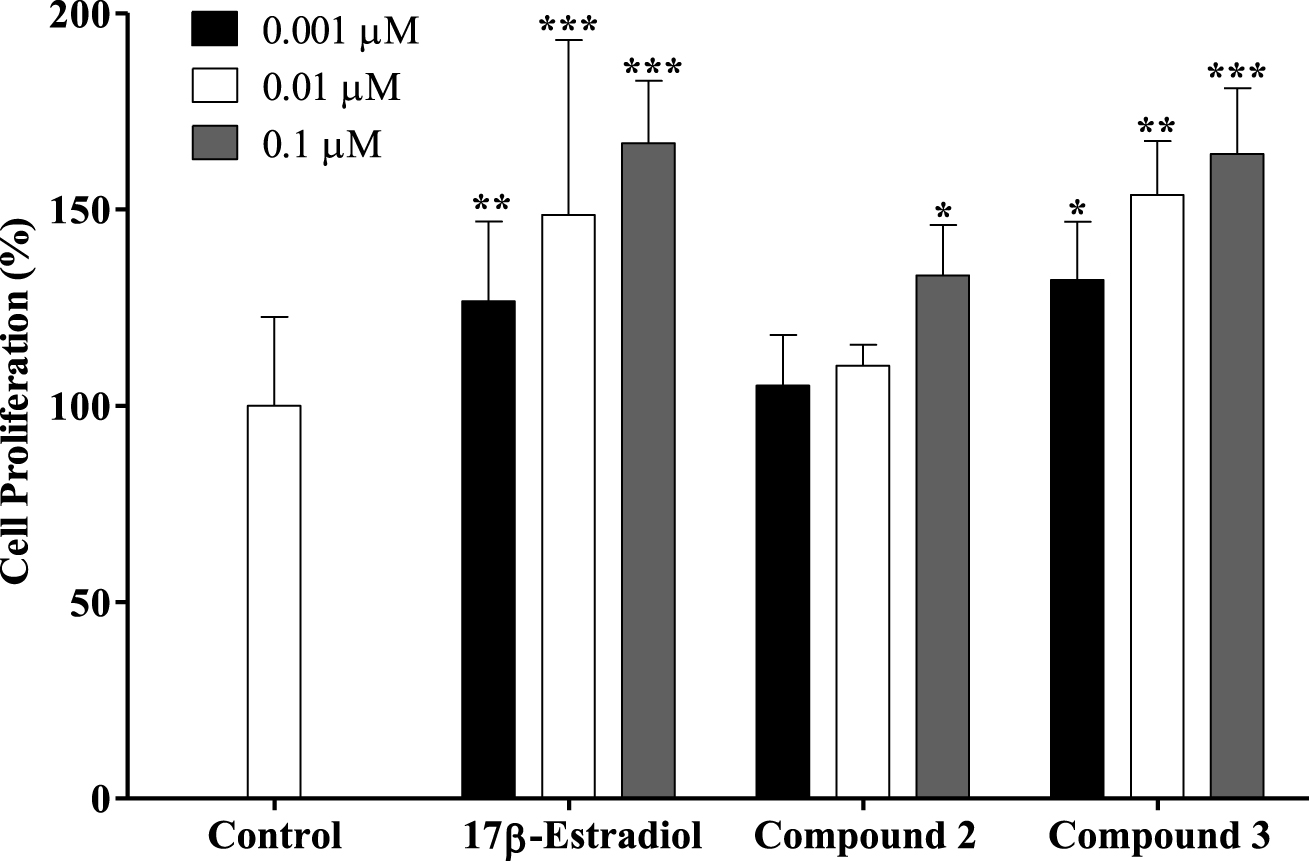
Prolifération des cellules T47-D sensibles aux œstrogènes après traitement par le 17ß-estradiol et les composés 2 et 3 pendant 24 h. Chaque barre représente la moyenne ± ET (provenant de deux expériences indépendantes). * p <0,05 vs contrôle; ** p <0,01 vs contrôle; *** p <0,001 vs contrôle.
Tableau 2.
Indice de sélectivité a des composés 1, 2 et 5-FU
a indice de sélectivité est le rapport de la CI 50 valeurs des traitements de cellules non tumorales (NHDF) et des cellules tumorales (MCF-7, T47-D, LNCaP, HepaRG et Caco-2). ND: non déterminé.
Un nouveau médicament candidat doit être dépourvu d'activité œstrogénique comme condition préalable à une utilisation dans le traitement du cancer. Afin d'étudier le profil œstrogénique potentiel des composés synthétisés ayant les activités antiprolifératives les plus pertinentes (stéroïdes 2 et 3), leur effet sur la croissance cellulaire a été mesuré sur les cellules T47-D du cancer du sein sensibles aux œstrogènes (ER +) dans un sérum-milieu de culture sans. Cette activité proliférative / œstrogène a été exprimée comme la différence entre la prolifération cellulaire (en pourcentage) causée par un composé donné et la prolifération cellulaire basale fixée à 100% (figure 3) [44, 45]. L'œstrogène naturel E2 a également été testé comme composé de référence. Comme prévu, E2 a eu un effet prolifératif sur les cellules T47-D à toutes les concentrations testées. Malheureusement, le composé 2 a également stimulé la prolifération cellulaire à 0,1 µM (133%) par rapport au contrôle négatif. Le composé 3 a également favorisé la prolifération cellulaire dans toutes les concentrations testées. Dans ce contexte, Palomino et al. [46], en utilisant des études de cristallographie aux rayons X et de modélisation moléculaire, ont montré que l' insaturation Δ9,11 dans E2 (affinité de liaison au récepteur, RBA = 1000) provoquait un aplatissement des anneaux B, C et D et par conséquent réduit le liaison à l'ER d'un cinquième (RBA = 196). Bien que la présence du Δ9,11 double liaison peut changer la conformation spatiale et réduire l'interaction avec le RE, elle n'a pas éliminé l'effet oestrogénique caractéristique de ces composés comme en témoignent nos résultats. De plus, Sakac et al. [47] ont confirmé l'effet œstrogénique du composé 3,6 ß-dihydroxyestra-1,3,5 (10), 9 (11)-tetraene-17ß-yl propionate en utilisant un dosage du poids utérin de rat immature (environ 73% de l'utérus prolifération par rapport au témoin). L'activité anti-œstrogène de ce composé a également été évaluée à l'aide d'une méthode anti-utérotrophique qui a montré un effet faible (3,22% d'effet antagoniste contre 62,80% pour le médicament de référence le tamoxifène) [15]. De nouveaux dérivés E1 modifiés en C-16 et C-17 ont été synthétisés par le groupe Alsayari [15], montrant une puissante inhibition de la croissance cellulaire stimulée par E2 et une affinité sélective élevée pour ERα . De plus, la 2-méthoxyestra-1,3,5 (10), 9 (11)-tetraen-17-one, qui possède une double liaison Δ9,11, a montré une activité œstrogénique et de bonnes affinités de liaison à ERα (4,09 µM) et ER ß (19,19 µM). Il a également été démontré qu'une chaîne latérale 2-bromoéthyle en C-3 et qu'une chaîne carbamoylbenzyle en C-16 éliminait l'activité œstrogénique résiduelle associée au noyau œstrogène [44, 48, 49]. Cependant, nos données ont montré que l'introduction du groupe benzylidène en C-16 n'était pas suffisante pour réduire l'effet oestrogénique de ce dérivé E1 sur les cellules T47-D.
3.2.2.
Évaluation de la survie cellulaire et de la distribution du cycle cellulaire
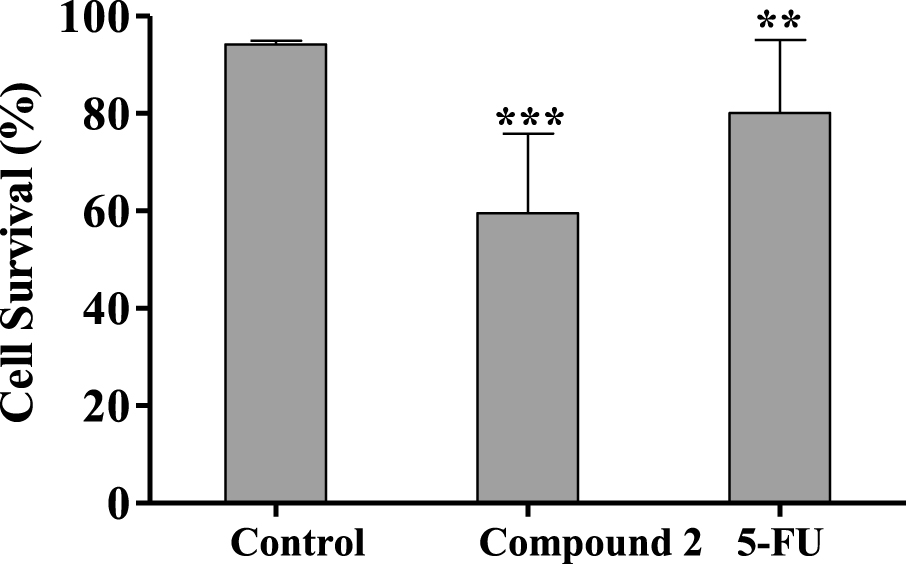
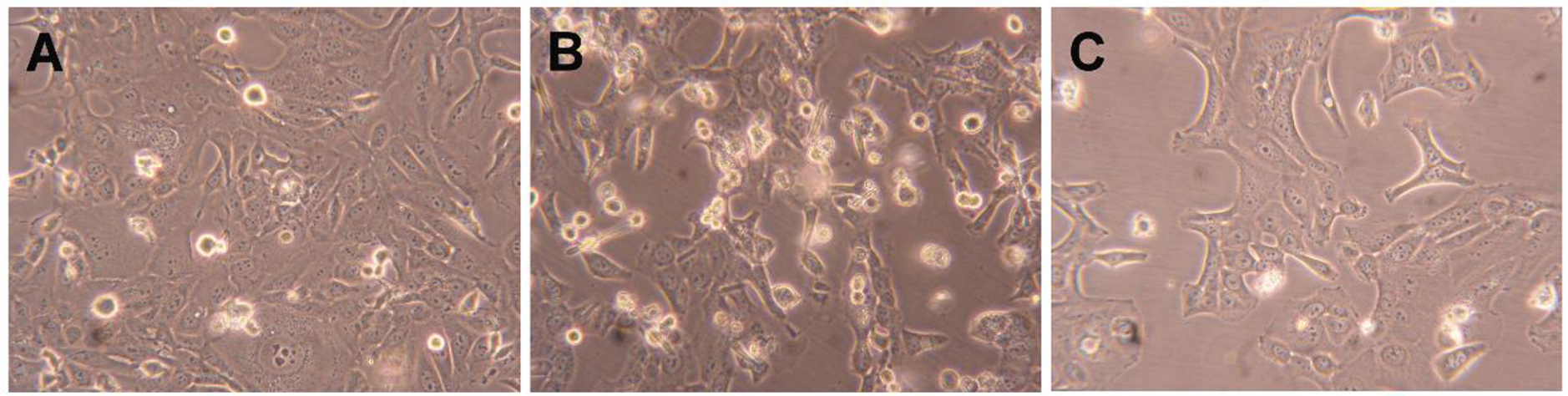
Le composé 2 a en outre été testé pour évaluer son mécanisme d'action possible par cytométrie en flux après coloration PI. Ce test a été réalisé dans des cellules HepaRG et du 5-FU a été utilisé comme contrôle positif. Dans cette lignée cellulaire, il a été observé que le composé 2 conduisait à une réduction de 34% de la viabilité cellulaire après 24 h de traitement (figure 4). Cet effet était plus élevé que celui causé par le 5-FU. En plus de cette étude de cytométrie en flux, les cellules ont également été observées avec un microscope optique (figure 5) et, après 24 h de traitement avec le composé 2, il était possible de voir de petites modifications dans les cellules HepaRG. Les cellules ont perdu leur forme, devenant plus arrondies.
Figure 4.
Pourcentage de cellules viables HepaRG après 24 h de traitement avec 50 µM de composé 2 évalué par cytométrie en flux à l'iodure de propidium (PI). Le contrôle correspond aux cellules non traitées et du 5-FU (50 µM) a été utilisé pour la comparaison. Le pourcentage de survie est le pourcentage de cellules dans R1 (cellules vivantes) par rapport au nombre total d'événements dans R1, R2(cellules mortes) et R3(cellules indéterminées). Chaque barre représente la moyenne ± ET (provenant de deux expériences indépendantes). ** p <0,01 vs contrôle; *** p <0,001 vs contrôle.
Figure 5.
Photographies de cellules HepaRG (A) traitées avec 50 µM de composé 2 (B) et 5-FU (C) pendant 24 h. Amplification de 100 ×.
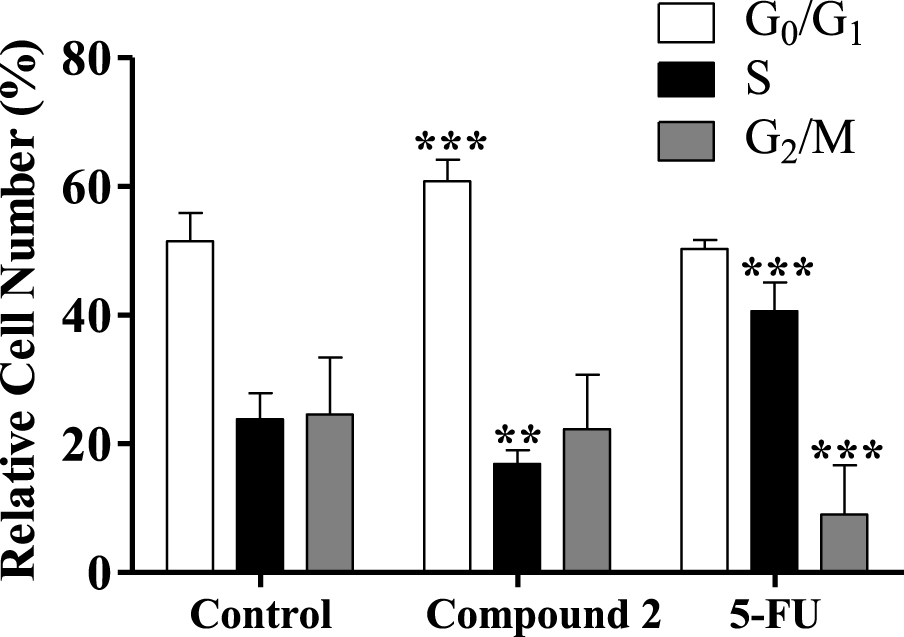
Certaines études ont montré que différents stéroïdes entraînaient un blocage du cycle cellulaire et inhibaient certaines enzymes importantes pour la régulation du cycle cellulaire. Par exemple, les 16ß-triazolyl-17 a-estradiol 3-benzyl éthers de la série 13 a-E2 ont montré un arrêt du cycle cellulaire G2/ M et une inhibition de la caspase [50]. De plus, de nouveaux dérivés de 3-benzyloxy-16-hydroxyméthylène-estradiol ont conduit à une accumulation de phase G1 et à un effet proapoptotique via l'élévation de la phase apoptotique sous-G1 sur les cellules MDA-MB-231 après 24 h de traitement (0,1–30 µM). De plus, on a observé que ces composés avaient une activité antimétastatique par inhibition de la phosphorylation de la kinase d'une manière dépendante de la concentration [51]. En tenant compte de ces informations, l'interférence du composé 2 dans la distribution du cycle cellulaire a été évaluée par cytométrie en flux. Fait intéressant, il a été constaté que le traitement avec le composé 2 (50 µM, 24 h) induisait un arrêt apparent du cycle cellulaire G0/G1 (figure 6), réduisant le pourcentage de cellules en phase S (réplication de l'ADN). L'arrêt du cycle cellulaire observé dans la phase G0/G1 peut être lié à l'interférence avec une ou plusieurs des nombreuses protéines qui participent aux mécanismes cellulaires hautement régulés qui retardent ou initient la réplication de l'ADN [52]. D'autres études seront nécessaires pour élucider quelles sont les voies de signalisation qui sont affectées et pour déterminer si d'autres mécanismes sont impliqués dans la cytotoxicité de ces composés.
Figure 6.
Analyse de la distribution du cycle cellulaire des cellules cancéreuses HepaRG après traitement avec le composé 2 (à 50 µM) pendant 24 h. Un contrôle négatif (cellules non traitées) et un contrôle positif [5-fluorouracile (5-FU), 50 µM] ont été inclus. L'analyse de la distribution du cycle cellulaire a été réalisée après coloration à l'iodure de propidium (PI) puis par cytométrie en flux. Chaque barre représente la moyenne ± ET (provenant de deux expériences indépendantes). ** p <0,01 vs contrôle; *** p <0,001 vs contrôle.
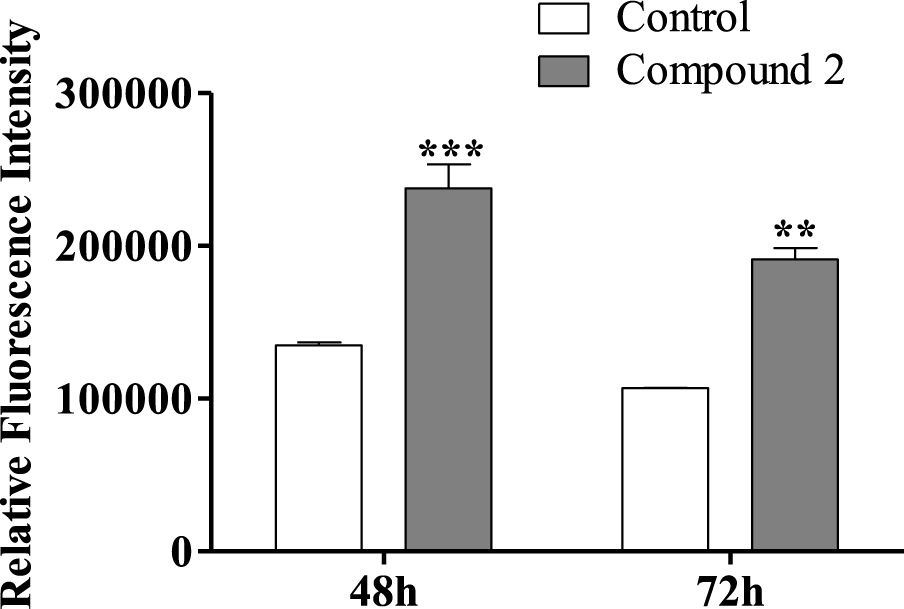
Concernant l'effet du composé 2 sur le cycle cellulaire HepaRG, nous avons également décidé d'étudier la prolifération cellulaire HepaRG après 72 h en utilisant un protocole adapté avec l'ester succinimidylique de carboxyfluorescéine, un colorant qui marque le cytoplasme cellulaire et est dilué lors de la division cellulaire [53]. Les cellules HepaRG traitées avec le composé 2 avaient un signal d'intensité plus élevé que les cellules témoins (figure 7), ce qui signifie qu'elles ont accumulé un nombre inférieur de divisions cellulaires et sont donc moins prolifératives. La diminution de la prolifération confirme que l'accumulation cellulaire dans G0/G1 est due à un arrêt dans le cycle cellulaire plutôt qu'à une progression plus rapide du cycle cellulaire à travers les phases S et G2/ M.
3.3. Études d'amarrage moléculaire
Les études d'amarrage moléculaire sont un déterminant dans la conception de médicaments basés sur la structure, car il est possible de prédire la conformation de liaison des ligands de petites molécules aux sites de liaison cibles appropriés, aux énergies de liaison et au mode de liaison dans la cible. Dans ce contexte, la caractérisation du comportement de liaison joue un rôle important dans la conception rationnelle des médicaments et nous aide à élucider les processus biochimiques fondamentaux [54]. Cette étude visait à évaluer l'existence d'interactions potentielles entre ces dérivés Δ9,11-estrone et des protéines connues pour interagir avec ces types de stéroïdes.
Figure 7.
Intensité relative de fluorescence de l'ester succinimidylique de carboxyfluorescéine (CFSE) des cellules HepaRG, évaluée par cytométrie en flux après traitement avec le composé 2 (50 µM) pendant 48 et 72 h. Chaque barre représente la médiane avec une plage de deux échantillons. ** p <0,01 vs contrôle; *** p <0,001 vs contrôle.
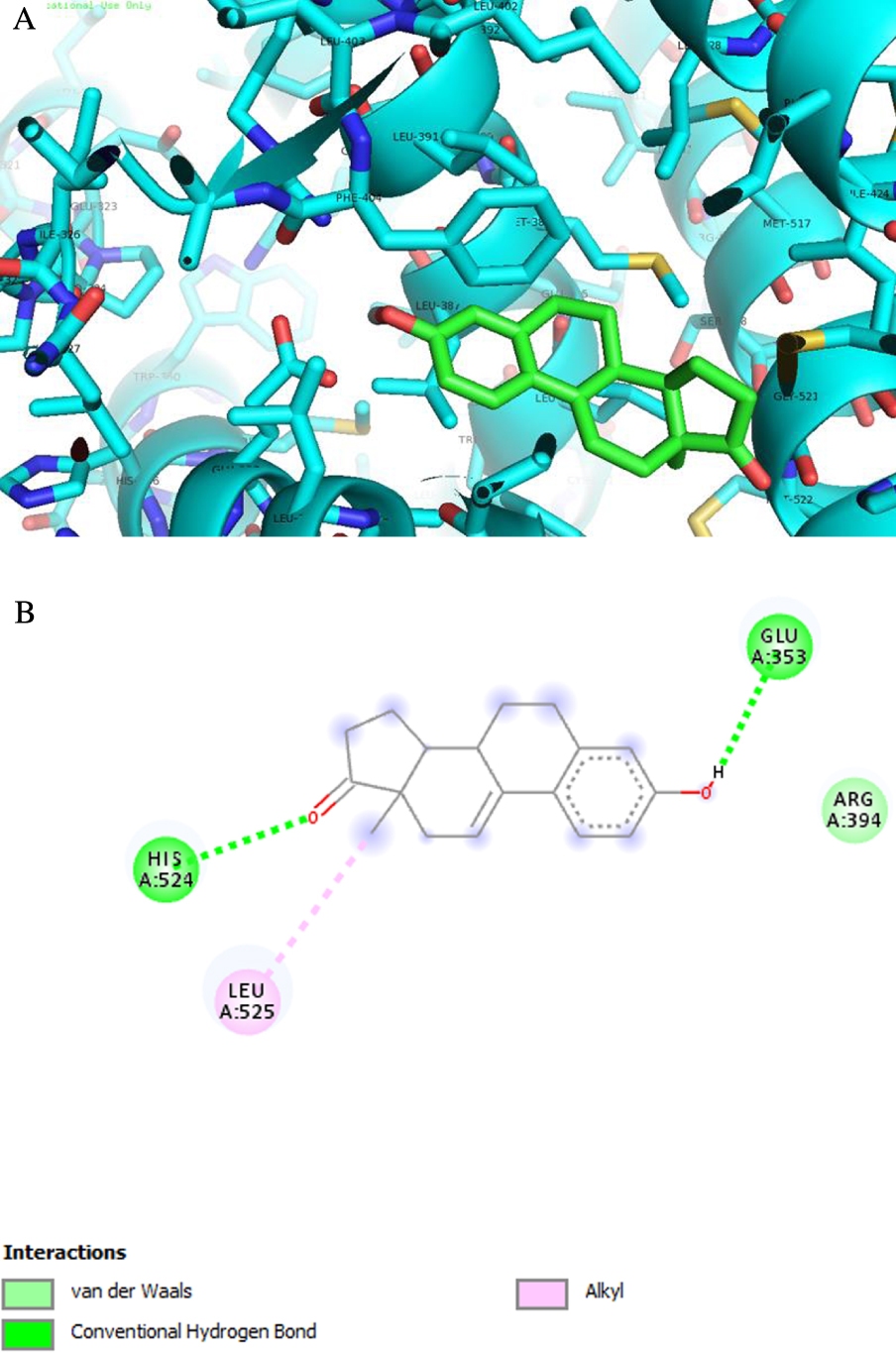
Figure 8.
Analyse des orientations de liaison prédites de ERα pour le composé le mieux classé, 2 (énergies de liaison inférieures aux énergies de re-docking). (A) Résultats d'amarrage 3D et (B) 2D montrant les principales interactions: liaisons H avec His 524 et Glu 353.
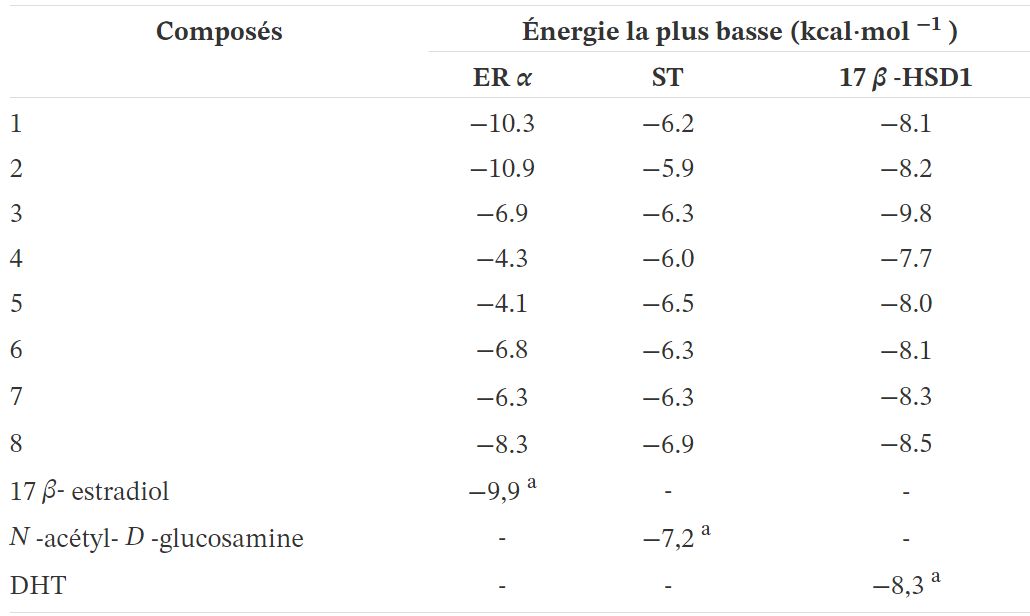
Tableau 3.
Énergies de liaison prévues des composés 1 à 8 calculées par rapport à ERα, ST et 17ß-HSD1 par AutoDockTools avec exécutable Vina. Les énergies de liaison du ligand présent dans les structures cristallines des rayons X ont été calculées par réamarrage
a Le RMSD entre les ligands réamarrés et les coordonnées de la structure cristalline des rayons X correspondantes était ≤2.
ERα est un facteur de transcription impliqué dans la régulation de nombreux processus physiologiques complexes chez l'homme. L'association entre l' activité ERα et le cycle cellulaire révèle que ce récepteur régule également la prolifération cellulaire ainsi que la résistance thérapeutique [55, 56, 57]. La ST et la 17ß-HSD de type 1 sont des enzymes également impliquées dans la prolifération cellulaire [58]. En fait, ST convertit le sulfate d'estrone en E1 et la 17ß-HSD de type 1 réduit la 17-cétone de l'androstane et les stéroïdes estrane en dérivés 17ß-hydroxylés correspondants, conduisant à une activité œstrogénique [59]. Par conséquent, leur dérégulation peut contribuer à la progression de plusieurs cancers hormono-dépendants. Les coordonnées structurales tridimensionnelles de ces trois récepteurs de protéines ont été obtenues à partir de la PDB, et l'ancrage moléculaire a été effectué à l'aide du programme AutoDock Vina. Pour valider la méthode d'amarrage, des simulations ont été réalisées et comparées à des ligands / médicaments cristallisés complexés avec les protéines respectives: toutes les simulations de re-docking de contrôle ont pu reproduire les géométries d'interaction ligand-protéine présentes dans les structures cristallines respectives avec un RMSD ≤2,0 Å. Tous les composés ont été ancrés pour ERα, ST et 17ß-HSD1 comme observé dans le tableau 3 . Fait intéressant, les résultats ont révélé que le composé 2 peut se lier à ERαà une énergie inférieure à celle du témoin 17ß-estradiol. D'après la figure 8 et concernant la cible ERα, le composé 2 présente deux liaisons hydrogène entre le groupe cétone en C-17 et Hist 524 et entre le groupe hydroxyle en C-3 et Glu 353. Ces interactions sont similaires à celles observées pour E2. Comme prévu, en ce qui concerne la 17ß-HSD1, l'énergie la plus faible par rapport au DHT témoin a été obtenue avec le composé 3, suivi du composé 8 . En fait, de nombreuses études ont été publiées impliquant des modifications en C-16 de E1 et C-2 de E2 pour développer 17 inhibiteurs ß-HSD 1 [12, 31, 48,49]. Le composé 3 a un groupe 16E-benzylidène en C-16, qui contribue à l'interaction avec la cible 17ß-HSD 1. Dans ce contexte, il a été démontré qu'un linker flexible en position C-16 donnait une meilleure inhibition de la 17ß-HSD1 que ceux avec un linker alcène rigide [60]. De plus, Bacsa et al. [29] ont montré que les dérivés 2 et/ou 4-halogénés de 13β- ou 13α-estrone conduisaient à une inhibition compétitive réversible des enzymes 17ß-HSD1 et ST. Concernant l'enzyme ST, aucun des composés étudiés n'a montré d'interaction pertinente avec cette cible. On sait que la présence d'un libre ou N-un groupe sulfamate non substitué (H2NSO2O–) est une condition préalable à une inhibition puissante et irréversible de la ST, comme le montrent des inhibiteurs comme EMATE [61].
4. Conclusion
En résumé, plusieurs estranes avec des modifications des anneaux A, C et D, y compris trois nouveaux dérivés Δ9,11-E1, ont été préparés dans des conditions de réaction douces. L'introduction d'une double liaison Δ9,11 et d'un groupe 16E-benzylidène dans E1 a augmenté l'activité cytotoxique sur les cellules cancéreuses du sein hormono-dépendantes (MCF-7 et T47-D) par rapport à E1. L'introduction de groupes 2,4-diiodo dans E1 semblait favoriser une sélectivité pour les cellules HepaRG. Cependant, la présence de groupes 2,4-diiodo et 2,4-dibromo dans Δ9,11-E1 ne semble pas avoir de bénéfice pour les activités antiprolifératives dans toutes les lignées cellulaires étudiées. Le résultat le plus prometteur a été observé avec Δ9,11-E1, qui présentait une activité antiproliférative pertinente contre les cellules cancéreuses HepaRG et présentait une cytotoxicité modérée sur des cellules humaines normales. Néanmoins, ce composé a également montré un effet œstrogénique sur les cellules T47-D à 0,1 µM, et l'analyse par cytométrie en flux a révélé un arrêt du cycle cellulaire à la phase G0/G1 des cellules HepaRG. Des études d'amarrage moléculaire ont estimé une forte interaction entre ce composé et ERα . En conclusion, la présence d'une double liaison Δ9,11 dans les dérivés estranaires peut être intéressante pour le développement de nouveaux agents antitumoraux intéressants.
Remerciements
Les auteurs reconnaissent le soutien apporté par les fonds FEDER à travers le POCI — COMPETE 2020 — Programme opérationnel Compétitivité et internationalisation dans l'axe I — Renforcement de la recherche, du développement technologique et de l'innovation (projet POCI-01-0145-FEDER-007491) et des fonds nationaux par FCT— Fondation pour la science et la technologie (Projet UID / Multi / 00709/2013). CC tient également à remercier le groupe du professeur Silvia Socorro (CICS-UBI) pour avoir fourni le DCC-FCS.
Conflit d'intérêt
Les auteurs confirment que le contenu de cet article n'a aucun conflit d'intérêts.
Données supplémentaires
Des informations complémentaires pour cet article sont disponibles sur le site Web de la revue sous https://doi.org/10.5802/crchim.17 ou auprès de l'auteur. Les détails expérimentaux et les protocoles de randomisation sont fournis.
Bibliographie
[1] (Cancer Research UK, Cancer Statistics Reports for the UK, 2019, https://www.cancerresearchuk.org/health-professional/cancer-statistics/worldwide-cancer/incidence#heading-One (accessed 21-10-2019))
[2] D. Archampong; H. Sweetland Surgery Volume 33 (2014), pp. 122-126
[3] J. F. Liu; M. Gordon; J. Veneris; F. Braiteh; A. Balmanoukian; J. P. Eder; A. Oaknin; E. Hamilton; Y. Wang; I. Sarkar; L. Molinero; M. Fassò; C. O’Hear; Y. G. Lin; L. A. Emens Gynecol. Oncol. Volume 154 (2019), pp. 314-322
[4] M. A. Abou-Salim; M. A. Shaaban; M. K. A. E. Hameid; Y. A. M. M. Elshaier; F. Halaweish Bioorg. Chem. Volume 85 (2019), pp. 515-533
[5] A. C. Groner; M. Brown J. Clin. Invest. Volume 127 (2017), pp. 1126-1135
[6] I. Kümler; A. S. Knoop; C. A. R. Jessing; B. Ejlertsen; D. L. Nielsen ESMO Open Volume 1 (2016), e000062 pages
[7] C. I. Lee; A. Goodwin; N. Wilcken Cochrane Database Syst. Rev. (2017) (CD011093, p. 1-54)
[8] J. A. R. Salvador; J. F. S. Carvalho; M. A. C. Neves; S. M. Silvestre; A. J. Leitão; M. M. C. Silva; M. L. Sá e Melo Nat. Prod. Rep. Volume 30 (2013), pp. 324-374
[9] L. G. A. Chuffa; L. A. Lupi-Júnior; A. B. Costa; J. P. de A. Amorim; F. R. F. Seiva Steroids Volume 118 (2017), pp. 93-108
[10] A. C. Society (Cancer Facts & Figures 2018, Atlanta, 2018)
[11] B. S. Kumar; D. S. Raghuvanshi; M. Hasanain; S. Alam; J. Sarkar; K. Mitra; F. Khan; A. S. Negi Steroids Volume 110 (2016), pp. 9-34
[12] M. Salah; A. S. Abdelsamie; M. Frotscher Mol. Cell. Endocrinol. Volume 489 (2019), pp. 66-81
[13] A. E. E. Amr; E. A. Elsayed; M. A. Al-Omar; H. O. B. Eldin; E. S. Nossier; M. M. Abdallah Molecules Volume 24 (2019), pp. 1-18
[14] R. Dutour; J. Roy; F. Cortés-Benítez; R. Maltais; D. Poirier J. Med. Chem. Volume 61 (2018), pp. 9229-9245
[15] A. Alsayari; L. Kopel; M. S. Ahmed; A. Pay; T. Carlson; F. T. Halaweish Steroids Volume 118 (2017), pp. 32-40
[16] M. Vosooghi; H. Yahyavi; K. Divsalar; H. Shamsa; A. Kheirollahi; M. Safavi; S. K. Ardestani; S. Sadeghi-Neshat; N. Mohammadhosseini; N. Edraki; M. Khoshneviszadeh; A. Shafiee; A. Foroumadi DARU J. Pharm. Sci. Volume 21 (2013), pp. 1-7
[17] R. Bansal; S. Guleria; S. Thota; R. W. Hartmann; C. Zimmer Chem. Pharm. Bull. Volume 59 (2011), pp. 327-331
[18] A. Stander; F. Joubert; A. Joubert Chem. Biol. Drug Des. Volume 77 (2011), pp. 173-181
[19] H. Chen; X. Liang; T. Sun; X. Qiao; Z. Zhou; Z. Li; C. He; H. Ya; M. Yuan Steroids Volume 134 (2018), pp. 101-109
[20] R. Maltais; A. Trottier; X. Barbeau; P. Lagüe; M. Perreault; J. Thériault; S. Lin; D. Poirier J. Steroid Biochem. Mol. Biol. Volume 161 (2016), pp. 24-35
[21] D. Milic; T. Kop; Z. Juranic; M. J. Gašic; B. Tinant; G. Pocsfalvi; B. A. Šolaja Steroids Volume 70 (2005), pp. 922-932
[22] T. R. Dias; M. G. Alves; S. P. Almeida; J. Silva; A. Barros; M. Sousa; B. M. Silva; S. M. Silvestre; P. F. Oliveira Steroid Biochem. Mol. Biol. Volume 154 (2015), pp. 1-11
[23] M. Jesus; A. P. J. Martins; E. Gallardo; S. Silvestre J. Anal. Methods Chem. (2016), pp. 1-16
[24] C. Canário; S. Silvestre; A. Falcão; G. Alves Curr. Med. Chem. Volume 25 (2018), pp. 660-686
[25] V. Brito; A. O. Santos; P. Almeida; S. Silvestre C. R. Chim. Volume 22 (2019), pp. 73-83
[26] R. B. Gabbard; L. F. Hamer; A. Segaloff Steroids Volume 37 (1981), pp. 243-255
[27] E. Stéphan; R. Zen; L. Authier; G. Jaouen Steroids Volume 60 (1995), pp. 809-811
[28] F. S. Alvarez; A. N. Watt J. Org. Chem. Volume 37 (1972), pp. 3725-3729
[29] I. Bacsa; B. E. Herman; R. Jójárt; K. S. Herman; J. Wölfling; G. Schneider; M. Varga; C. Tömböly; T. L. Rižner; M. Szécsi; E. Mernyák J. Enzyme Inhib. Med. Chem. Volume 33 (2018), pp. 1271-1282
[30] P. C. B. Page; F. Hussain; N. M. Bonham; P. Morgan; J. L. Maggs; B. K. Park Tetrahedron Volume 47 (1991), pp. 2871-2878
[31] D. Poirier; H. J. Chang; A. Azzi; R. P. Boivin; S. X. Lin Mol. Cell. Endocrinol. Volume 248 (2006), pp. 236-238
[32] D. Ispán; E. Szánti-Pintér; M. Papp; J. Wouters; N. Tumanov; B. Zsirka; Á. Gömöry; L. Kollár; R. Skoda-Földes European J. Org. Chem. Volume 2018 (2018), pp. 3236-3244
[33] D. M. Tanenbaum; Y. Wang; S. P. Williams; P. B. Sigler Proc. Natl Acad. Sci. USA Volume 95 (1998), pp. 5998-6003
[34] F. G. Hernandez-Guzman; T. Higashiyama; W. Pangborn; Y. Osawa; D. Ghosh J. Biol. Chem. Volume 278 (2003), pp. 22989-22997
[35] J. A. Aka; M. Mazumdar; C. Q. Chen; D. Poirier; S. X. Lin Mol. Endocrinol. Volume 24 (2010), pp. 832-845
[36] W. H. W. Lunn; E. Farkas Tetrahedron Volume 24 (1968), pp. 6773-6776
[37] W. Brown; J. W. A. Findlay A. B. Chem. Commun. (1968), p. 10-11
[38] H. Guo; H. Wu; J. Yang; Y. Xiao; H.-J. Altenbach; G. Qiu; H. Hu; Z. Wu; X. He; D. Zhou; X. Hu Steroids Volume 76 (2011), pp. 709-723
[39] R. Bansal; S. Guleria Steroids Volume 73 (2008), pp. 1391-1399
[40] J. A. Egan; C. N. Filer Appl. Radiat. Isot. Volume 71 (2013), pp. 68-71
[41] L. Lista; A. Pezzella; A. Napolitano; M. D’Ischia Tetrahedron Volume 64 (2008), pp. 234-239
[42] M. Cushman; H.-M. He; J. A. Katzenellenbogen; C. M. Lin; E. Hamel J. Med. Chem. Volume 38 (1995), pp. 2041-2049
[43] C. Bézivin; S. Tomasi; F.L.-L. Dévéhat; J. Boustie Phytomedicine Volume 10 (2003), pp. 499-503
[44] D. Ayan; R. Maltais; J. Roy; D. Poirier Mol. Cancer Ther. Volume 11 (2012), p. 2096-104
[45] F. Cortés-Benítez; J. Roy; R. Maltais; D. Poirier Bioorganic Med. Chem. Volume 25 (2017), pp. 2065-2073
[46] E. Palomino; M. J. Heef; J. P. Horwitz; L. Polin; S. C. Brooks Mol. Biol. Volume 50 (1994), pp. 75-84
[47] M. N. Saka?; K. M. Penov Gaši; M. Popsavin; E. A. Djurendi?; S. Andri?; R. M. Kova?evi? Collect. Czechoslov. Chem. Commun. Volume 70 (2005), pp. 479-486
[48] R. Maltais; D. Ayan; D. Poirier ACS Med. Chem. Lett. Volume 2 (2011), pp. 678-681
[49] Y. Laplante; C. Cadot; M.-A. Fournier; D. Poirier Bioorganic Med. Chem. Volume 16 (2008), pp. 1849-1860
[50] E. Mernyák; I. Kovács; R. Minorics; P. Sere; D. Czégány; I. Sinka; J. Wölfling; G. Schneider; Z. Újfaludi; I. Boros; I. Ocsovszki; M. Varga; I. Zupkó J. Steroid Biochem. Mol. Biol. Volume 150 (2015), pp. 123-134
[51] I. Sinka; A. Kiss; E. Mernyák; J. Wölfling; G. Schneider; I. Ocsovszki; C.-Y. Kuo; H.-C. Wang; I. Zupkó Eur. J. Pharm. Sci. Volume 123 (2018), pp. 362-370
[52] P. Icard; L. Fournel; Z. Wu; M. Alifano; H. Lincet Trends Biochem. Sci. Volume 44 (2019), pp. 490-501
[53] M. Sánchez-Sánchez; L. Hernández-Linares; M. G. Escobar; H. López-Muñoz; E. Zenteno; G. Fernández-Herrera; M. A. Guerrero-Luna; J. Carrasco-Carballo Molecules Volume 21 (2016), E1533 pages
[54] D. B. Kitchen; H. Decornez; J. R. Furr; J. Bajorath Nat. Rev. Drug Discov. Volume 3 (2004), pp. 935-949
[55] H. R. Lee; K. A. Hwang; M. A. H. Park; B.-R. Yi; E. B. Jeung; K. C. Choi Int. J. Mol. Med. Volume 29 (2012), pp. 883-890
[56] A. J. Begam; S. Jubie; M. J. Nanjan Bioorg. Chem. Volume 71 (2017), pp. 257-274
[57] Y. Miki; E. Iwabuchi; K. Ono; H. Sasano; K. Ito Int. J. Mol. Sci. Volume 19 (2018), 3173 pages
[58] K. M. C. Cornel; C. Krakstad; B. Delvoux; S. Xanthoulea; B. Jori; M. Y. Bongers; G. F. J. Konings; L. F. S. Kooreman; R. F. Kruitwagen; H. B. Salvesen; A. Romano Mol. Cell. Endocrinol. Volume 442 (2017), pp. 51-57
[59] A. H. Payne; D. B. Hales Endocr. Rev. Volume 25 (2004), pp. 947-970
[60] G. M. Allan; H. R. Lawrence; J. Cornet; C. Bubert; D. S. Fischer; N. Vicker; A. Smith; H. J. Tutill; A. Purohit; J. M. Day; M. F. Mahon; M. J. Reed; B. V. L. Potter J. Med. Chem. Volume 49 (2006), pp. 1325-1345
[61] L. W. L. Woo; B. Leblond; A. Purohit; B. V. L. Potter Bioorg. Med. Chem. Volume 20 (2012), pp. 2506-2519