
1. Introduction
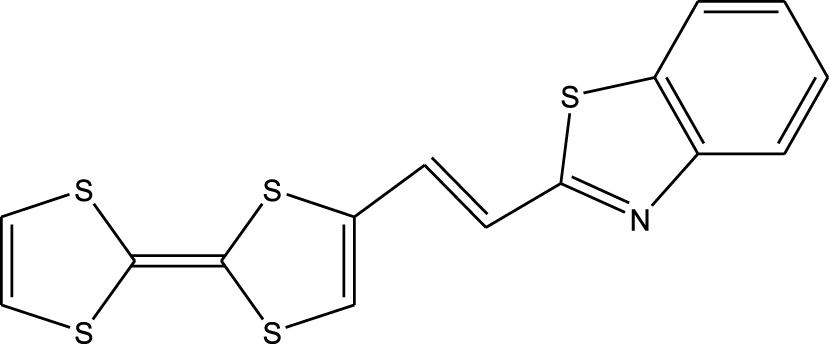
Le tétrathiafulvalène (TTF) et ses dérivés, en tant qu'oléfines substituées, sont connus des données expérimentales [1] et théoriques [2] pour avoir d'excellentes propriétés de donneur. Ceci est également confirmé par les dérivés oxydés stables mono-cationiques (TTF + ) et di-cationiques (TTF 2+ ) pouvant être obtenus via deux processus d'oxydation séquentiels et réversibles. Ces capacités permettent des applications utiles dans la chimie des matériaux [3], des conducteurs [4] et des supraconducteurs [5], des adduits au C 60 [6,7], des polymères conducteurs [8], des matériaux pour l’optique non linéaire (OLN) [9, 10], éponges cationiques [11], aimants organiques ferromagnétiques [12], cristaux liquides [13], dendrimères [14], rotaxanes moléculaires et caténanes [15]. Récemment, les systèmes moléculaires à base de TTF avec des groupes donneurs d'électrons (D) et accepteurs (A) ont suscité un intérêt considérable en tant que dyades (D– A). Ceux-ci sont utilisés comme commutateurs de fluorescence tels que le TTF-PPD (2,5-diphényl-1,3,4-oxadiazole), qui présente une forte fluorescence et peut être utilisé comme antenne pour la photoexcitation [16], les capteurs chimiques, la rectification moléculaire, et applications photovoltaïques et OLN [17, 18, 19, 20, 21, 22]. Cet article vise principalement une description complète de la géométrie moléculaire et des vibrations moléculaires de la molécule tétrathiafulvalène-1,3-benzothiazole (TTF– CH = CH– BTA), déjà discutée dans la littérature [23, 24, 25] et présentée dans le schéma 1. Dans cette molécule, le TTF a été choisi comme donneur en raison des propriétés précitées de ses dérivés et du cycle 1,3-benzothiazole, qui est une partie essentielle des matériaux organiques photofonctionnels.
En outre, les études spectroscopiques ultraviolettes visibles (UV– Vis) ainsi que l’orbitale moléculaire occupée la plus élevée - l’orbitale moléculaire inoccupée la plus basse (HOMO-LUMO) peuvent expliquer les effets de transfert de charge dans la molécule actuelle. En particulier, les propriétés de polarisation et de transfert de charge ont été déterminées en calculant les charges du tenseur de polarisabilité atomique (APT), le potentiel électrostatique moléculaire (MEP) et les paramètres optiques non linéaires. Ce dernier, qui comprend le moment dipolaire, la polarisabilité et la première hyperpolarisabilité de la molécule, a été établi afin d'acquérir des connaissances plus approfondies sur la relation entre l'architecture moléculaire et la réponse non linéaire, à l'instar des travaux d'autres auteurs [26, 27]. Enfin, la méthode orbitale de liaison naturelle (NBO) a été appliquée pour analyser la stabilité de la molécule résultant de l'interaction hyperconjugative et de la délocalisation de charge.
2. Méthodes de calcul
Tous les calculs ont été effectués avec le package Gaussian 16 [28]. La méthode de la théorie fonctionnelle de la densité (DFT) a été utilisée avec la fonctionnelle B3LYP (la fonction d'échange non local à trois paramètres de Becke avec la fonction de corrélation Lee– Yang– Parr) [29, 30] et l' ensemble de base de Pople [31] 6-31G * *. Les optimisations géométriques ont été réalisées en phase gazeuse et les minima et ont été confirmées par des calculs de fréquence; ceux-ci concordaient de manière satisfaisante avec les données structurelles expérimentales. En conséquence, les énergies et autres propriétés physico-chimiques pourraient être comparées en toute sécurité. Les spectres d'absorption UV– Vis simulés de la molécule TTF– CH = CH– BTA ont été obtenus par la méthode TD-DFT (B3LYP) selon les lignes suivies par d'autres auteurs [32]. Les énergies HOMO et LUMO correspondantes ont ensuite été utilisées pour estimer certains paramètres de réactivité chimique globale, tels que le potentiel chimique (μ), l'électronégativité (χ), l'indice d'électrophilicité (ω) et la dureté chimique (η) et la douceur (S) [33, 34]. De plus, les charges partielles APT, la carte du potentiel électrostatique moléculaire (MEP) et le moment dipolaire de TTF– CH = CH– BTA ont été calculés de manière similaire. Les analyses NBO et OLN de structures spécifiques ont également été réalisées pour les espèces optimisées.
3. Résultats et discussion
3.1. Géométrie moléculaire
Les paramètres structuraux optimisés de la molécule TTF– CH = CH– BTA dans le schéma 1 sont présentés dans le tableau 1. La structure moléculaire optimisée représentée par le logiciel Chemcraft est illustrée à la figure 1. Les valeurs calculées optimisées ont été comparées à celles d'une structure cristalline [25]. L'unité 1,3-benzothiazole (BTA) est conjuguée au TTF par un espaceur acétylénique, qui acquiert éventuellement des caractéristiques éthyléniques. Ce composé a été signalé en 2010 par Fujiwara et ses collègues. Il a été considéré comme un conducteur commutable utile pour la conversion photoélectrique de matériaux sélectionnés [23, 24]. En fait, des mesures avec un monocristal de TTF– CH = CH– BTA suggéraient l'existence de photocourants le long des deux unités TTF et BTA empilées [25]. Un transfert d'électrons intermoléculaire pourrait être photoinduit avec la formation d'états séparés de charge.
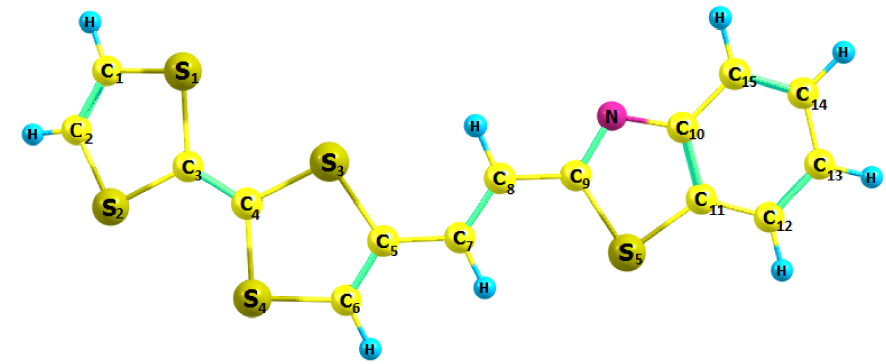
En général, les observations du tableau 1 indiquent un bon accord entre les paramètres structurels calculés et les données expérimentales. Des différences mineures étaient probablement dues à des valeurs optimisées obtenues dans une phase gazeuse isolée et à des valeurs expérimentales dans des cristaux. En bref, les distances moyennes des liaisons C=C, C–C et C–S calculées au niveau DFT / B3LYP / 6-31G ** étaient respectivement de 1,370, 1,419 et 1,744 Å, ce qui était légèrement supérieur aux valeurs expérimentales moyennes (1,347 , 1,384 et 1,733 Å, respectivement). Dans l'unité BTA, les distances de liaison C(9)–N et C(10)=N de 1,380 et 1,303 Å, respectivement, sont proches des valeurs expérimentales (1,395 et 1,313 Å, respectivement). De plus, il y a une certaine déviation de l'atome S par rapport au plan de la bicyclette dans l'unité TTF, comme le confirment les angles dièdres du côté TTF (C(1)–S(1)–C(3)–S(2) = −9,3, C(2)–S(2)–C(3)–S(1) = 9,3 , C(5)–S(3)–C(4)–S(4)=13.5 et C(6)–S(4)–C(4)–S(3) = −13.0), ce qui ne permet pas à la molécule d'être rigoureusement planaire.
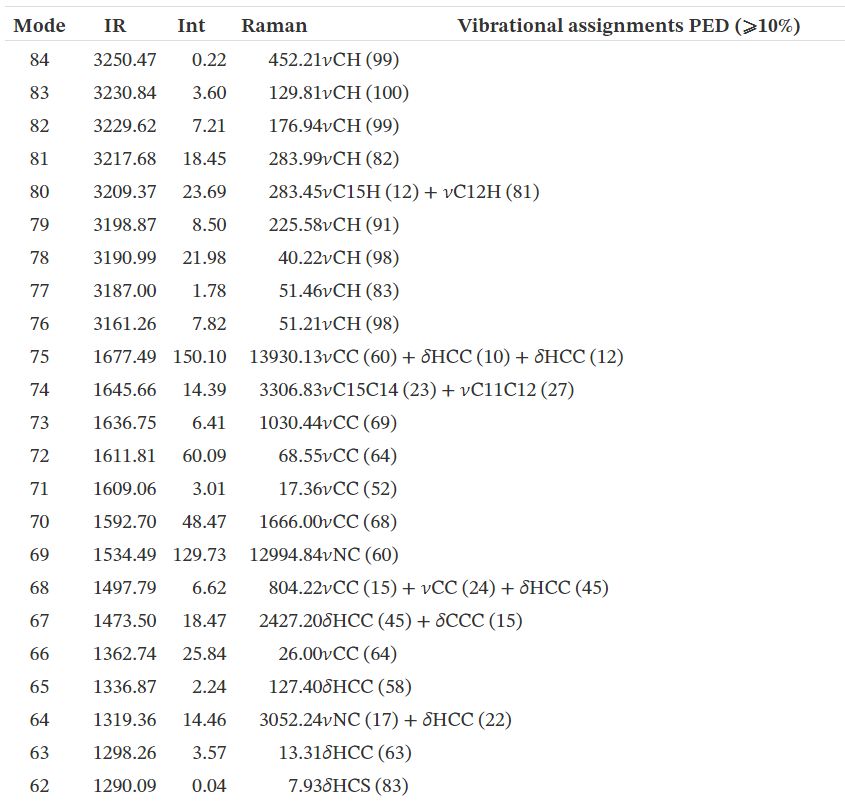
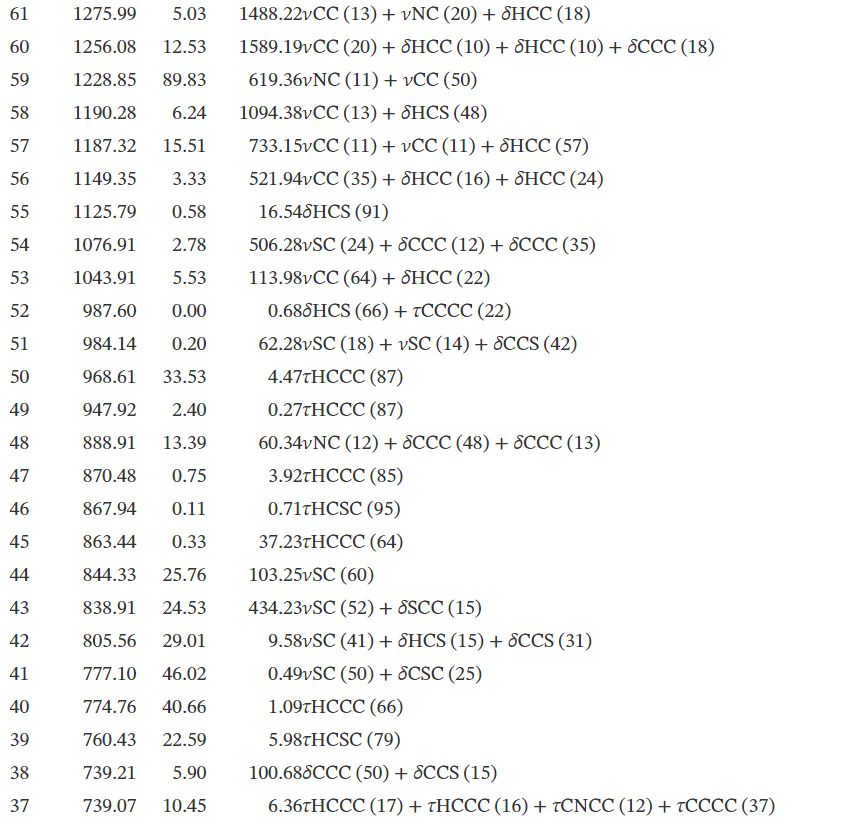
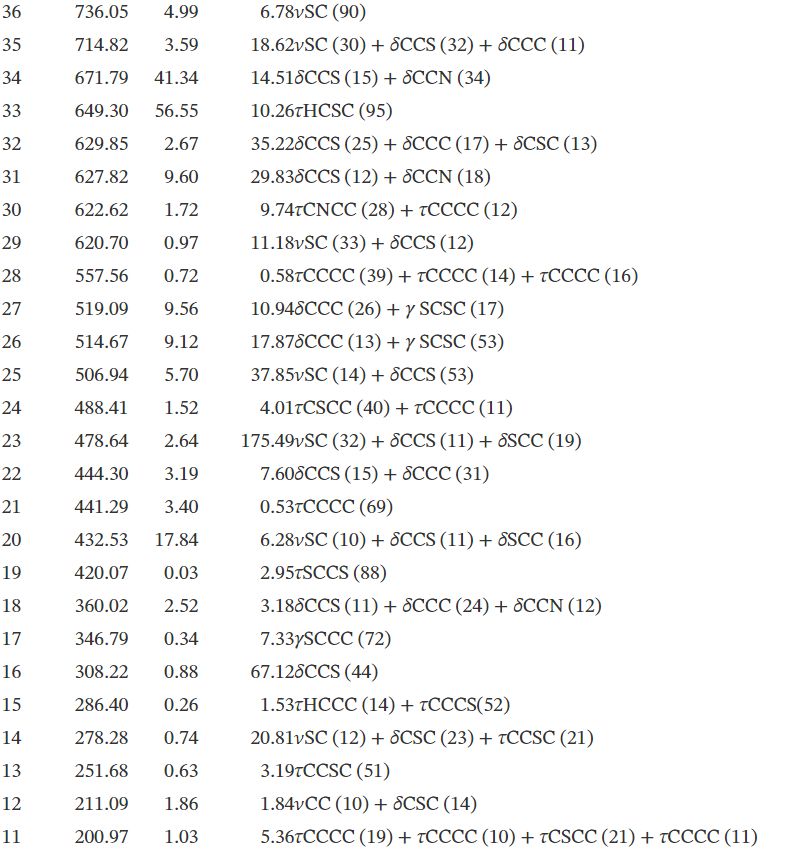

3.2. Analyse vibrationnelle
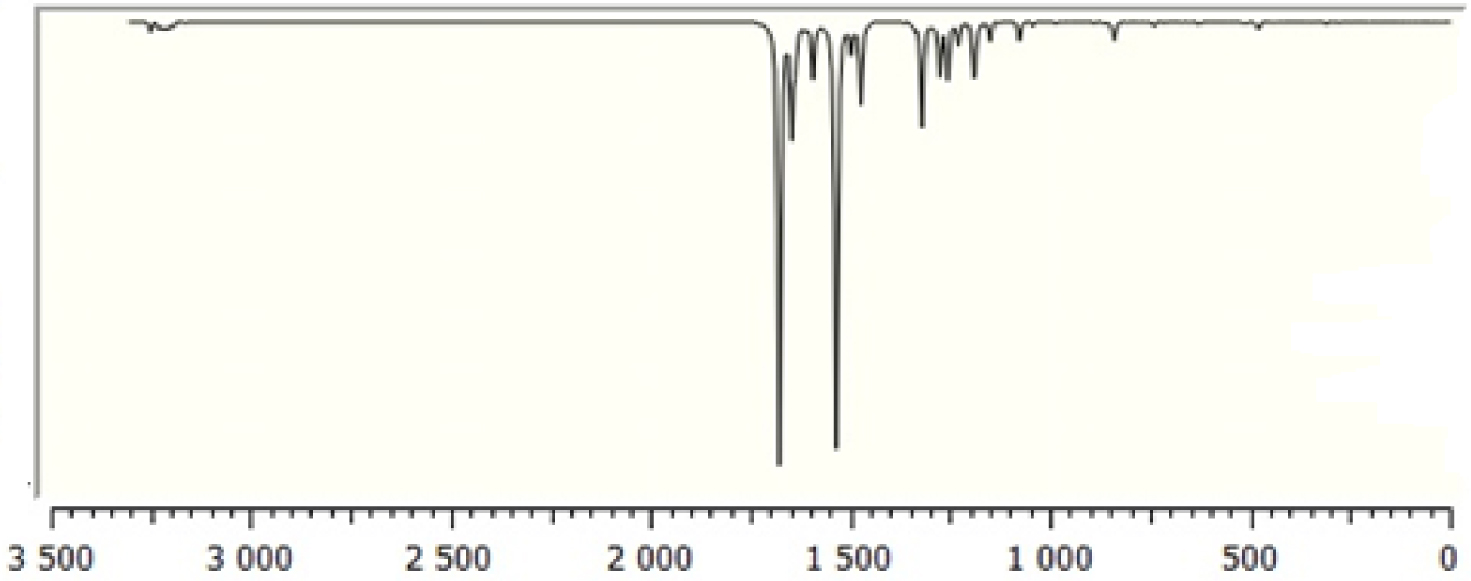
La molécule se compose de 30 atomes, ce qui implique 84 modes normaux de vibrations, qui sont actifs à la fois dans l'absorption infrarouge (IR) et la diffusion Raman. Les nombres d'onde vibrationnels fondamentaux de TTF– CH = CH– BTA ont été calculés par la méthode DFT avec B3LYP comme ensemble de base 6-31G **. Les résultats des activités IR (nombres d'ondes vibrationnelles et intensités IR) et Raman pour la structure optimisée sont répertoriés dans le tableau #tab22. Les spectres IR et Raman calculés sont affichés dans les figures 2 et 3. Les affectations vibrationnelles ont été réalisées sur la base du calcul de la distribution d'énergie potentielle à l'aide du programme «Analyse de la distribution d'énergie vibratoire» (VEDA).
3.2.1. Vibrations TTF
En général, les composés non aromatiques présentent des vibrations d'étirement C– H dans la région inférieure à 3000 cm-1 alors que les valeurs supérieures à 3000 cm-1 sont typiques des composés aromatiques [3535]. Dans la présente étude, les modes calculés à 3250, 3231 et 3230 cm-1 sont identifiés comme des modes d'étirement C– H symétriques et asymétriques, tandis que les nombres d'ondes observés se situent à 3099 cm-1 . Les données théoriques et expérimentales des modes d'étirement C– H de la partie TTF montrent une très bonne corrélation avec la littérature [36]. Les modes d'étirement C = C se trouvent dans la plage de nombres d'onde 1565–1495 cm-1 . Les modes d'étirement C– S sont observés dans la plage de nombres d'onde 935–734 cm-1et sont en bon accord avec l'aspect général des modes d'étirement ν(C– S).
3.2.2. Vibrations BTA
En raison de la présence de l'unité BTA, le spectre du composé implique les bandes correspondant à la flexion C– H, C– C, C– C– C et H– C– C ainsi qu'à la torsion C– C– C– C les vibrations. Les quatre liaisons C– H dans BTA correspondent aux vibrations à 3209, 3187, 3199 et 3218 cm-1 . Les vibrations de flexion C– H dans le plan sont liées aux vibrations d'étirement C– C et aux vibrations des structures moléculaires aromatiques se produisant dans la région de 500 à 100 cm-1 [37]. Des vibrations de flexion C– H dans le plan ont été obtenues à 1256, 1187, 1678, 1337, 1319, 1276, 1256, 1149, 1474, 1044, 1678, 1298, 1498 et 1190 cm-1 . Les vibrations d'étirement C– C des structures aromatiques se produisent fortement entre 1650 et 430 cm-1 [37]. Dans cette étude, les vibrations d'étirement C– C ont été calculées à 1646, 1498, 1149, 1276, 1363 et 1046 cm-1.
3.2.3. Vibrations C=C
Une vibration d'étirement conjuguée C=C a été rapportée à 1600 cm-1 [35]. Dans la présente étude, les vibrations d'étirement C– C sont calculées à 1678 cm-1 .
3.3. Charges atomiques (APT)
Les charges atomiques dans TTF–CH=CH–BTA ont été dérivées en calculant le tenseur polaire atomique (APT). Ce tenseur est calculé comme la somme des tenseurs de charge et de flux de charge qui permettent de construire un modèle de flux charge–charge [38]. Les charges atomiques correspondantes de TTF–CH=CH–BTA sont indiquées dans le tableau 3.
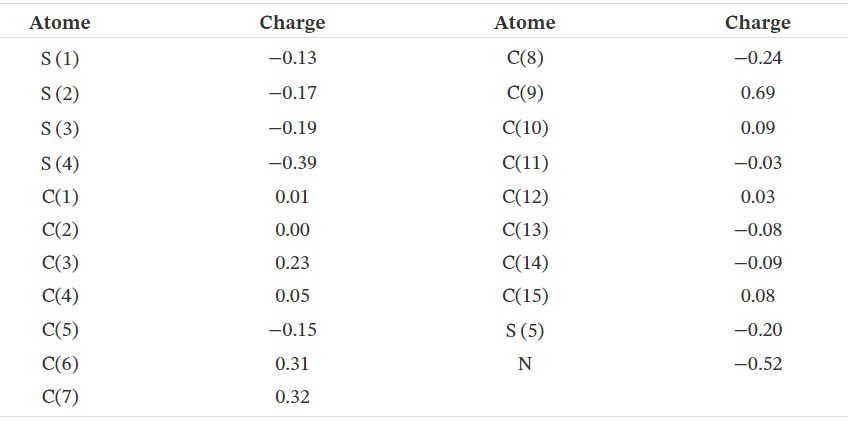
Les calculs de charge atomique jouent un rôle important dans les études de mécanique quantique des systèmes moléculaires car ils sont utilisés pour expliquer les moments dipolaires moléculaires, les structures électroniques et les polarisabilités et donc les réactivités chimiques. De l'analyse de la charge atomique de TTF–CH=CH–BTA, il ressort que l'atome d'azote dans l'unité BTA a la charge négative maximale de -0,52 e. Les atomes de soufre sont également chargés négativement avec des valeurs de -0,13, -0,20 et -0,39 e. L'atome C (9) du motif benzothiazole est le plus chargé positivement (+0,69 e); la plupart des autres atomes de C sont également chargés positivement à l'exception de C (5), C (8), C (14) et C (13), qui portent des charges négatives de -0,15, -0,24, -0,09 et -0,08 e, respectivement.
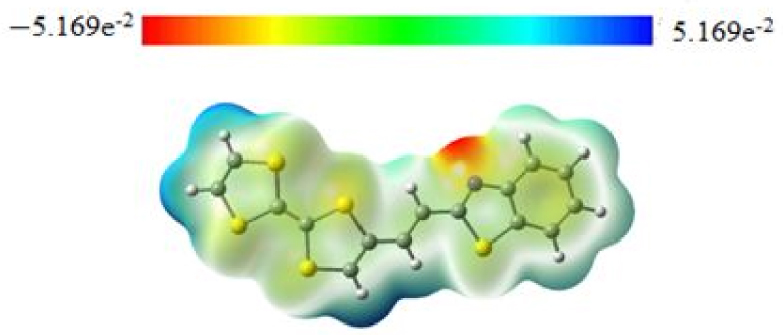
3.4. Potentiel électrostatique moléculaire
Le MEP a été généralement utilisé pour tenir compte des interactions électrostatiques dans une variété de systèmes chimiques [39]. À tout point \(\vec{r}\), le potentiel moléculaire électrostatique \(V (\vec {r})\) correspond à la force agissant sur une charge de test positive (un proton) située à \(\vec {r}\) à travers le nuage de charge généré par les électrons et les noyaux de la molécule. Pour le système actuel, les valeurs \(V (\vec {r})\) ont été calculées comme décrit précédemment en utilisant l'équation [40]
\begin{equation}\tag{1} V(\vec {r}) = \sum _{A}\frac {Z_{A}}{(\vec {R}_{A}-\vec {r})} - \int \frac {\rho (\vec {r}')}{(\vec {r}'-\vec {r})}d\vec {r}'. \end{equation} Dans (1), \(Z_A\) est la charge du noyau A, situé en \(\vec {R} _ {A}\), et \({\rho} (\vec {r} ')\) est la fonction de densité électronique de la molécule.Le MEP de TTF–CH=CH–BTA utilisant la géométrie optimisée DFT-B3LYP / 6-31G ** a été calculé, et sa carte de surface est illustrée à la figure 4. Cette figure montre les valeurs de potentiel électrostatique en utilisant un schéma de code couleur. La valeur la plus négative est caractérisée par le rouge, qui suggère les sites privilégiés pour une éventuelle attaque électrophile. Les régions les plus chargées positivement apparaissent en bleu foncé, ce qui indique à son tour les sites favoris pour une attaque nucléophile. Les limites déterminées sont -5,169e-2 (rouge le plus profond) et + 5,169e-2 (bleu le plus profond), avec l'échelle intermédiaire des couleurs passant du rouge à l'orange, au jaune, au vert et au bleu dans l'ordre, comme le montre la figure 4. Le potentiel le plus négatif est concentré autour de l'atome d'azote unique, tandis que les potentiels positifs sont périphériques, en particulier au niveau des atomes H externes de l'unité TTF. La positivité est un peu moins marquée pour les atomes H de l'unité BTA. En conclusion, le MEP suggère principalement une éventuelle attaque électrophile sur l'atome d'azote avec une certaine possibilité également pour deux atomes TTF S. Inversement, une base forte peut avoir une chance d'éliminer en tant que proton l'un des deux atomes H du linker HC=CH ou en d'autres termes de transformer le groupement éthylénique correspondant en un anion vinylidène du côté du substituant TTF.
3.5. Orbitales moléculaires frontière
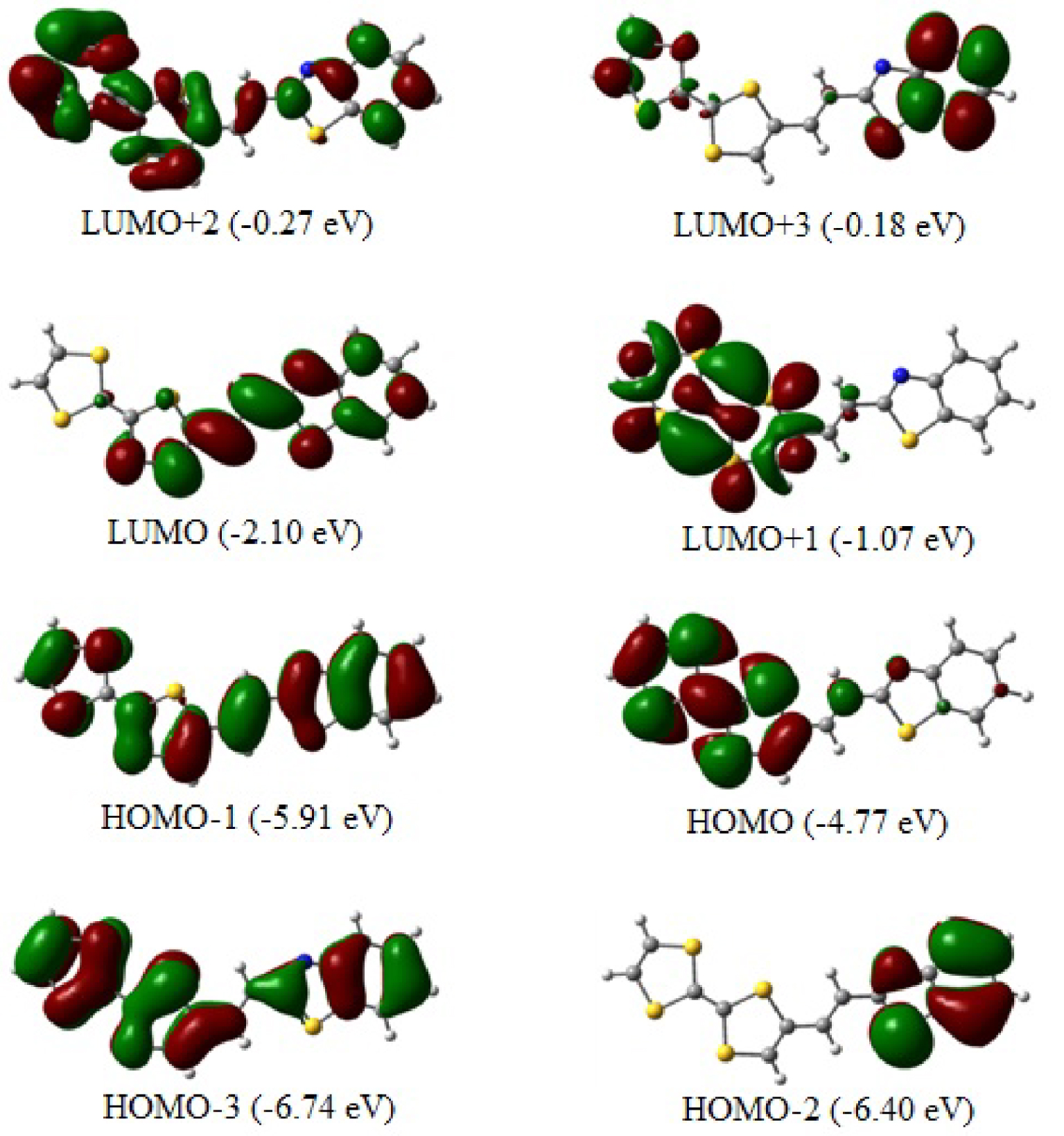
Les vues de dessus des orbitales moléculaires frontalières optimisées TTF–CH=CH–BTA (de HOMO-2 à LUMO+3) sont représentées sur la figure 5 avec le HOMO et le LUMO jouant un rôle important dans le transfert de charge intramoléculaire (ICT). Les caractéristiques topologiques de ces niveaux sont importantes pour interpréter la stabilité cinétique (et par conséquent, la réactivité chimique potentielle) ainsi que d'autres propriétés telles que les propriétés optiques [41, 42]. Comme prévu, toutes les orbitales moléculaires impliquées ont un caractère π qui s'étend sur la molécule plane globale.
L'écart HOMO–LUMO calculé est aussi grand que 2,67 eV. Le fragment TTF contribue principalement au HOMO. Le LUMO est principalement centré sur le lieur éthylénique, bien qu'il y ait une certaine contribution pπ résiduelle des atomes à ses côtés gauche et droit. A l'inverse, le caractère π du HOMO, qui est distribué dans toute la molécule TTF–CH=CH–BTA, indique comment la délocalisation d'électrons peut impliquer l'ensemble du système également grâce à la nature π des liens. Le gap énergétique HOMO–LUMO est relativement important et la transition électronique correspondante implique une redistribution significative de la densité électronique dans la molécule. En particulier, l'écart semble expliquer l'origine de l'épaulement de bande détecté dans les spectres UV-Vis expérimentaux précédemment rapportés [21]. Plus précisément, la forme donnée du spectre corrobore l'ICT entre le donneur TTF et les fragments de benzothiazole, ce dernier représentant le côté accepteur de la molécule.
3.6. Analyse spectrale UV–Vis
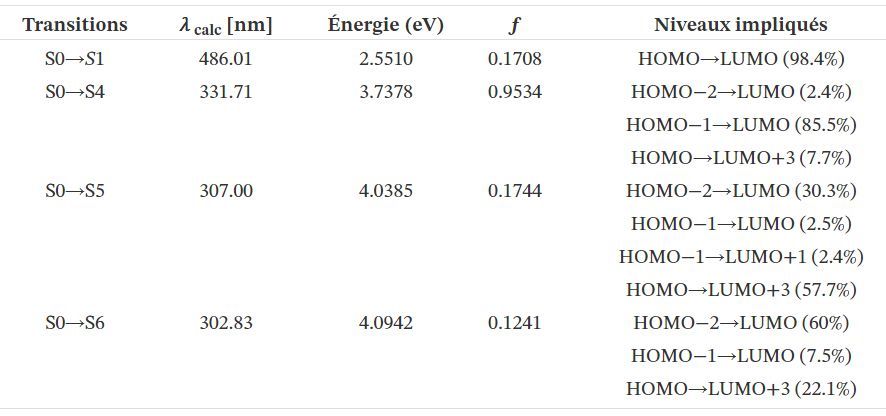
Les énergies verticales d'excitation ont été calculées en utilisant la méthode DFT en fonction du temps (TD-DFT) [43] afin de définir la nature des transitions électroniques et l'affectation des bandes observées dans les spectres d'absorption UV–Vis. Les résultats des calculs sont résumés dans le tableau 4 et les spectres simulés sont présentés à la figure 6.
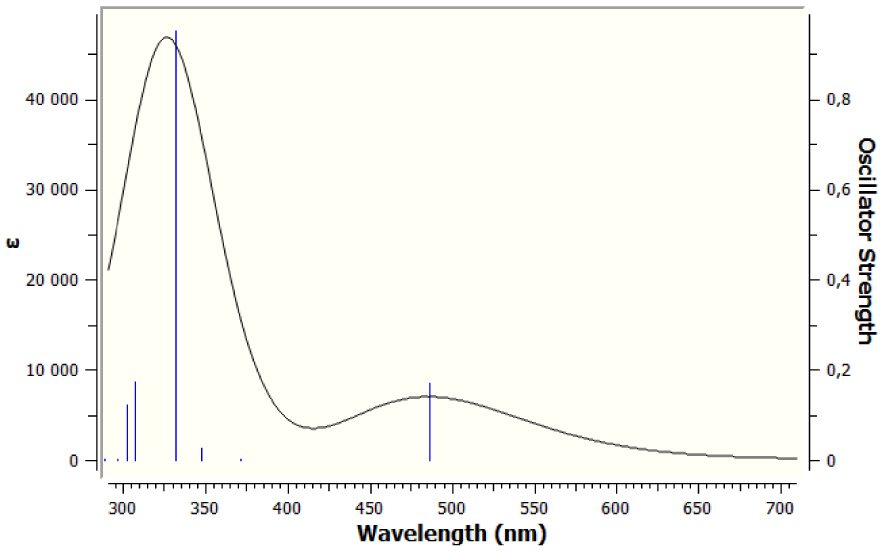
D'après les résultats du tableau 4, il ressort que la transition S0 → S1 la plus pertinente qui détermine le premier état excité se produit à 486,01 nm. Il s'agit essentiellement du transfert d'électrons HOMO–LUMO, étant donné que son poids est évalué à 98,4%. De plus, compte tenu de la composition des niveaux, la transition met en évidence la manière dont la cible de l'ICT en TTF–CH=CH–BTA est dirigée vers le groupement TTF.
La transition la plus intense suivante, avec une constante de force d'oscillation de 331,71 nm, se produit de l'état fondamental au quatrième état excité (S0 → S4). Dans ce cas, sa composante principale de 85,5% implique la transition électronique du HOMO-1 au LUMO, tandis qu'un autre 7,7% implique le HOMO et le LUMO + 3.
3.7. Descripteurs globaux de réactivité
Les descripteurs de réactivité globale tels que le potentiel d'ionisation (I), l'affinité électronique (A), l'électronégativité (χ), la dureté chimique (η), la douceur chimique (S), les valeurs de potentiel chimique (μ) et l'indice d'électrophilicité (ω) ont été examinés sur la base sur l'optimisation de TTF–CH=CH–BTA en phase gazeuse. Les paramètres fournissent des informations sur la stabilité chimique de la molécule. Les descripteurs numériques de réactivité, qui sont principalement basés sur les énergies HOMO–LUMO, sont résumés dans le tableau 5.

Le potentiel d'ionisation (I) et l'affinité électronique (A) selon le théorème de Koopmans peuvent être définis par les équations suivantes [44]:
\begin {equation}\tag{2} I = {-}E_{\mathrm {HOMO}}; \quad A = {-}E_{\mathrm {LUMO}}. \end {equation} L'électronégativité (\({\chi}\)), la dureté chimique globale (η) et le potentiel chimique électronique (μ) sont exprimés par [45] \begin {eqnarray} & {\mu}=(\partial E / \partial N) _ {v (\vec {r})}=- {\chi} & \tag{3} \end{eqnarray} \begin{eqnarray} & 2 {\eta}=(\partial \mu / \partial N) _ {v (\vec {r})}=(\partial \chi / \partial N) _ {v (\vec {r})}=(\partial ^ {2} E / \partial N ^ {2}) _ {v (\vec {r})}. \quad & \tag{4} \end{eqnarray} Dans les formules, \(E\), N et \(\vec{r}\) sont respectivement l'énergie totale du système, le nombre d'électrons et le potentiel externe. En outre, les relations suivantes connectent I et A avec les paramètres de Eq. (3) et (4) [44]: \begin {equation} \tag{5} {\mu}={-} ({I} + {A}) / 2, \quad {\eta}=({I} - {A}) / 2. \end{equation} L' indice global de douceur chimique (S) et d'électrophilicité ( \({\omega}\)) sont définis comme suit: \begin{equation} \tag{6} {S}=1 / {\eta}, \quad {\omega}=\mu ^ {2} / 2 {\eta}. \end{equation} Le potentiel d'ionisation (\(I\)) est défini comme la quantité d'énergie nécessaire pour éliminer un électron d'une molécule. De plus, une énergie d'ionisation élevée indique une stabilité élevée et donc une inertie chimique, tandis qu'une faible énergie d'ionisation suggère une propension de la molécule à la réactivité. L'affinité électronique (A) est définie comme l'énergie libérée lorsqu'un électron est ajouté à une molécule neutre et donc une grande valeur (A) indique la tendance de la molécule à conserver ses électrons. Un potentiel chimique négatif (μ) indique la stabilité moléculaire ou la difficulté de la molécule à se décomposer en ses propres éléments. La dureté (η) caractérise la résistance du nuage d'électrons moléculaires à la déformation lors de petites perturbations. Un grand écart d'énergie HOMO-LUMO indique une molécule dure avec une faible polarisabilité et de faibles activités chimiques et biologiques mais une forte sensibilité cinétique, tandis qu'un petit écart d'énergie HOMO-LUMO indique une molécule molle avec une polarisabilité élevée et des activités chimiques et biologiques mais une faible sensibilité cinétique. L'indice d'électrophilie global (ω) d'une molécule est une mesure de son énergie de stabilisation suite à l'ajout d'une charge électronique externe ou de sa résistance pour échanger l'électron avec le système [46]. Pour le composé du titre, les valeurs calculées du potentiel d'ionisation, de l'affinité électronique, de l'électronégativité, du potentiel chimique, de la dureté, de la douceur et de l'électrophilicité étaient respectivement de 4,77, 2,10, 3,43, -3,43, 1,33, 0,75 et 4,44 eV. Notez que l'énergie d'ionisation élevée et le potentiel chimique négatif (μ) indiquent la stabilité, c'est-à-dire que la molécule ne se décompose pas spontanément en ses éléments.3.8. Descripteurs de réactivité locale
L'indicateur de réactivité le plus important est la fonction de Fukui f(r), proposée en 1984 par Parr et Yang [47]. La fonction au point r est la dérivée de la densité électronique par rapport à la variation du nombre d'électrons N, étant donné le potentiel externe V(r):
\begin{equation} \tag{7} f (r)={\Bigg (\frac {\partial \rho (r)} {\partial N} \Bigg)} _ {{V _ {(r)}}} . \end{equation} Puisque le nombre d'électrons N est une variable discrète, les dérivés ioniques mono-positifs et mono-négatifs de \({\rho} (r)\) en ce qui concerne N sont apparus. En appliquant une approximation aux différences finies à l'équation ci-dessus, nous obtenons trois définitions des fonctions de Fukui en fonction de la densité électronique totale.
- Pour les attaques nucléophiles, \begin{eqnarray} f_ {k} ^ {+} (r) &=& {\left (\frac {\partial \rho (r)} {\partial N} \right)} _ {v} ^ {+ } \tag{8} \end{eqnarray} \begin{eqnarray} {} & {}={} & {} [\rho _ {N + 1} (r) - \rho _ {N} (r)] \tag{9} \end{eqnarray} \begin {eqnarray} {} & {}={} & {} [q_ {k} (N + 1) -q_ {k} (N)]. \label {eq10} \end {eqnarray}
- Pour les attaques électrophiles, \begin {eqnarray} f_ {k} ^ {-} (r) &=& {\left (\frac {\partial \rho (r)} {\partial N} \right)} _ {v} ^ {- } \tag{11} \end{eqnarray} \begin {eqnarray} {} & {}={} & {} [\rho _ {N} (r) - \rho _ {N-1} (r)] \tag{12} \end {eqnarray} \begin {eqnarray} {} & {}={} & {} [q_ {k} (N) -q_ {k} (N-1)]. \tag{13} \end {eqnarray}
- Pour les attaques radicales, \begin {eqnarray} f_ {k} ^ {0} (r) &=& {\left (\frac {\partial \rho (r)} {\partial N} \right)} _ {v} ^ {0 } \tag{14} \end {eqnarray} \begin {eqnarray} &=& 1/2 [\rho _ {N + 1} (r) - \rho _ {N-1} (r)] \tag{15} \end {eqnarray} \begin {eqnarray} &=& 1/2 [q_ {k} (N + 1) -q_ {k} (N-1)]. \tag{16} \end {eqnarray}
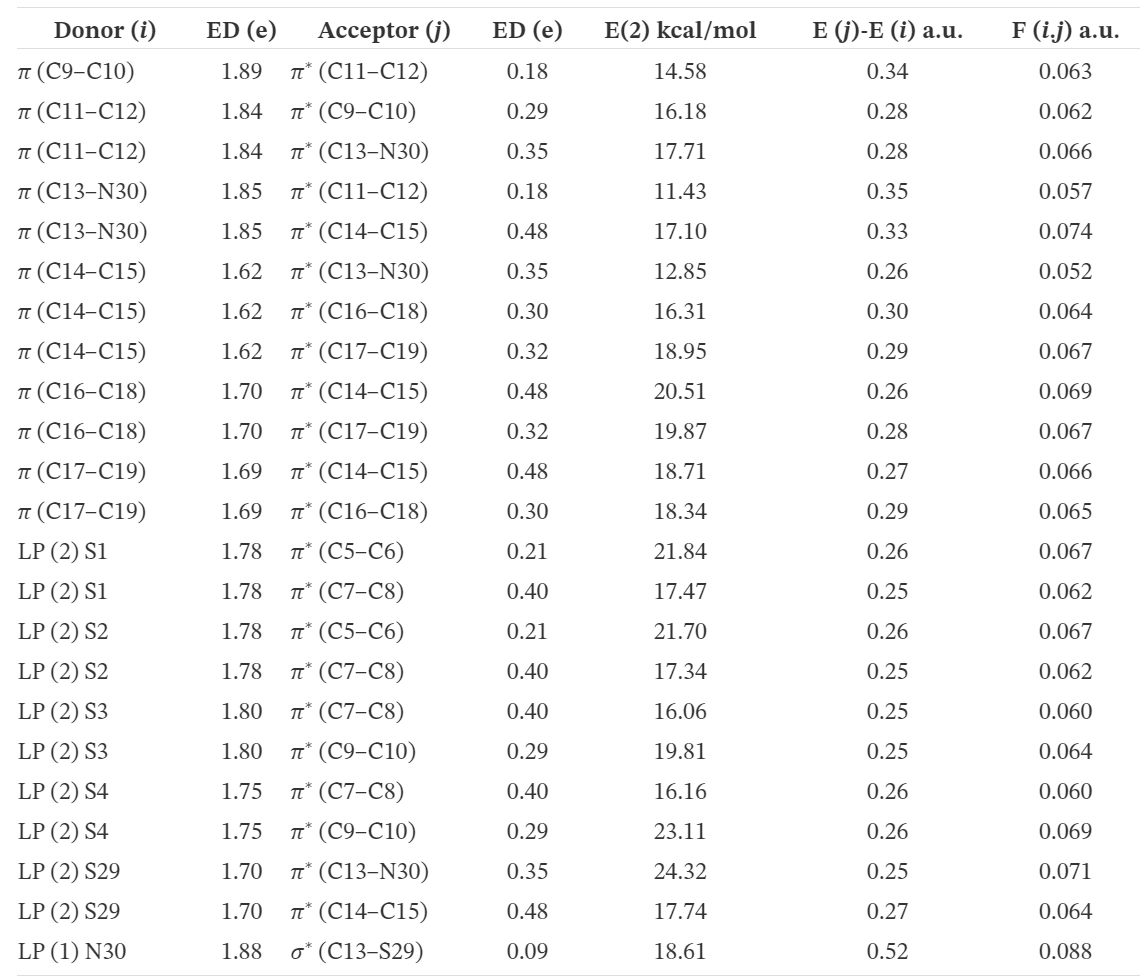
Les paramètres de réactivité locale du tableau 6 indiquent que l'atome d'azote est le site le plus réactif pour les attaques de types électrophiles et radicaux libres, tandis que le C(8) éthylénique est le plus sujet à une attaque nucléophile.
3.9. Analyse orbitale des liaisons naturelles
L'analyse NBO est une méthode fiable pour examiner les propriétés de transfert de charge et la nature de liaison intra et intermoléculaire d'une molécule donnée. La délocalisation d'électrons π implique qu'une orbitale de valence de Lewis donnée (donneur) subit une diminution d'occupation en raison de la densité électronique, qui est déplacée vers une autre région de la molécule (accepteur). La théorie appliquée de la perturbation du second ordre montre une certaine diminution d'énergie associée à cet effet. Pour chaque donneur (NBO ( i )) et accepteur (NBO ( j )), la force de l'interaction de délocalisation ou de l'énergie de stabilisation ( E (2) ) associée à la redistribution d'électrons entre les côtés donneur et accepteur peut être formulée comme la seconde- ordre de réduction d'énergie [48, 49, 50]:
\begin{equation} E ^ {(2)}={-} q_ {i} \frac {({F_ {ij}}) ^ {2}} {\varepsilon _ {i} - \varepsilon _ {j} }. \end{equation} Ici, \(q_ {i}\) est l'occupation orbitale du donneur; εi et εj sont les énergies orbitales des orbitales NBO donneur et accepteur, respectivement; Fij est l'élément de matrice de Fock ou Kohn–Sham hors diagonale. Les données extraites de l'analyse de la théorie des perturbations du second ordre de la matrice de Fock de TTF–CH=CH–BTA sont présentées dans le tableau 7.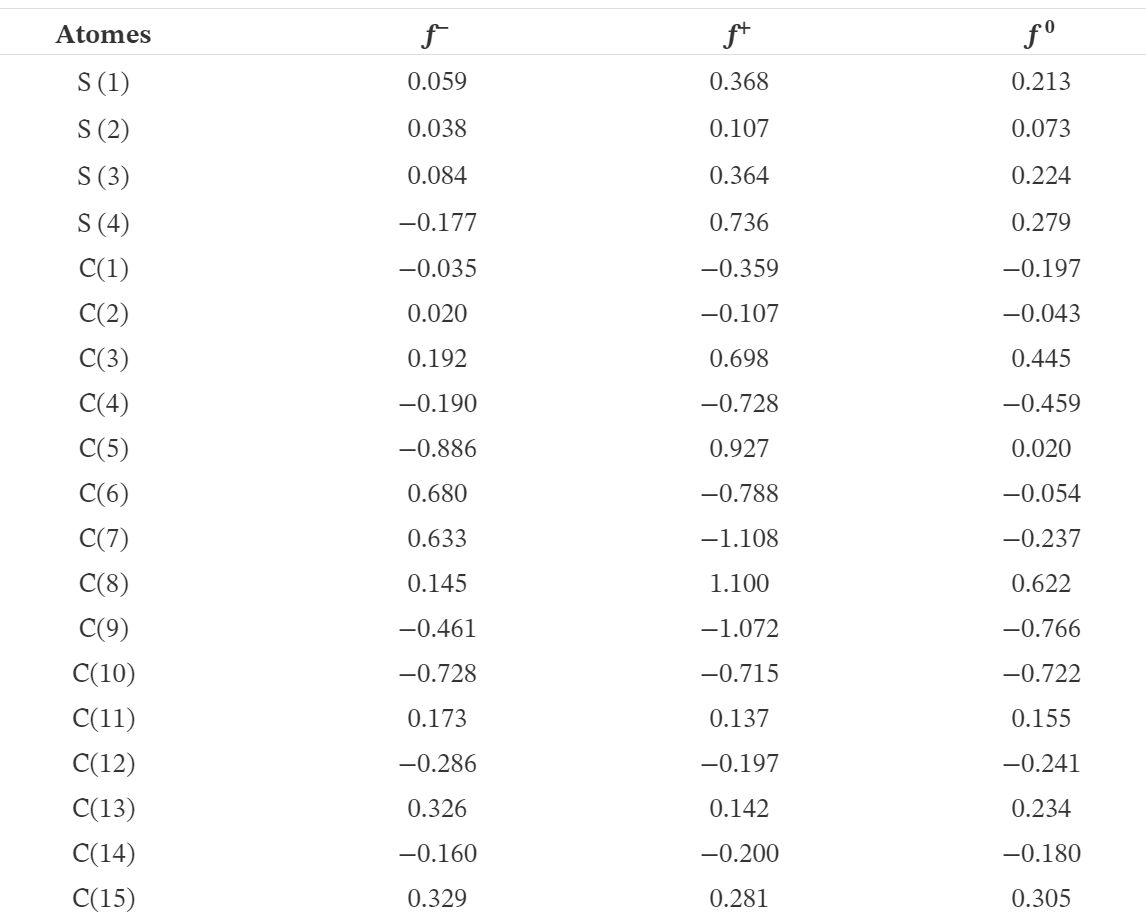
Les résultats ci-dessus suggèrent que l'interaction la plus importante dans la molécule est celle entre la seule paire S(5) avec le composant π* (C9–N) et S(4) avec π* (C5–C6). Dans ces cas, les énergies de stabilisation correspondantes sont respectivement de 24,32 et 23,11 kcal / mol. En conséquence, un transfert d'énergie important est observé dans ces interactions par paires. En particulier, les transferts d'électrons dans les niveaux π* (C12–C13) et π* (C7–C8) induisent des énergies de stabilisation significatives de 18,95 et 14,58 kcal / mol, respectivement.
3.10. Propriétés optiques non linéaires
Les propriétés optiques non linéaires d'une molécule se sont avérées être une ligne directrice importante pour établir la structure moléculaire et l'assemblage des matériaux organiques. Ces propriétés jouent un rôle dans les technologies actuelles telles que la communication et les processus informatiques [51, 52]. Les effets OLN résultent des interactions des champs électromagnétiques avec divers milieux qui modifient les caractéristiques des champs incidents tels que la phase, la fréquence, l'amplitude ou d'autres aspects de propagation [53]. Les effets OLN sont importants pour assurer les fonctions clés des décalages de fréquence, de la modulation optique, de la commutation optique, de la logique optique, de la mémoire optique et des technologies émergentes dans les télécommunications, le traitement du signal et les interconnexions optiques [54,55, 56, 57]. Par conséquent, le DFT a été largement utilisé pour étudier les matières organiques OLN [58, 59, 60, 61, 62].
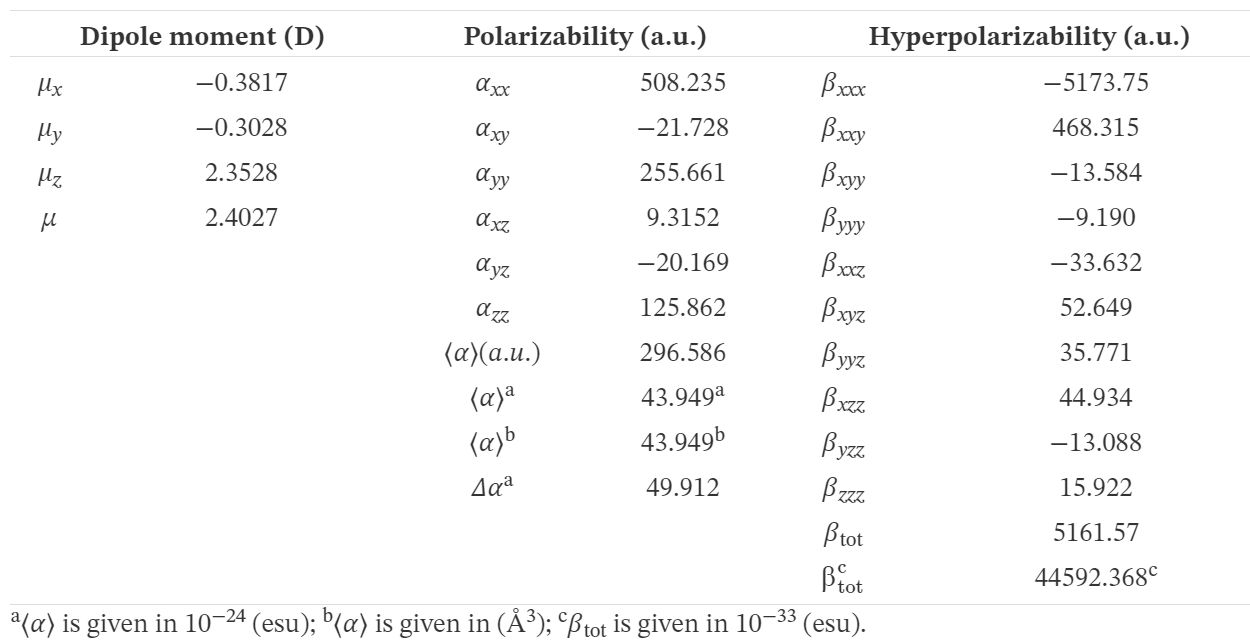
Les propriétés, telles que le moment dipolaire (μ), la polarisabilité (αij ) et la première hyperpolarisabilité (βtot ), sont liées à des propriétés optiques non linéaires. Cette méthode DFT utilisée à nos fins a déjà fait ses preuves dans l'étude des matières organiques avec OLN. Nos valeurs dérivées sont résumées dans le tableau 8. Les équations utilisées pour dériver la valeur du moment dipolaire total (μtot ), la polarisabilité isotrope moyenne (⟨α⟩), l'anisotropie de la polarisabilité Δα et le premier ordre de l'hyperpolarisabilité (βtot ), respectivement, sont les suivantes [63]:
\begin{eqnarray}\tag{18} \mu _ {\mathrm {tot}} &=& \sqrt {{\mu _ {x}} ^ {2} + {\mu _ {y}} ^ {2} + {\mu _ {z}} ^ {2}} \end {eqnarray} \begin {eqnarray}\tag{19} \langle \alpha \rangle &=& \frac {1} {3} \mathop {(} \alpha _ {xx} + \alpha _ {yy} + \alpha _ {zz} \mathop {)}\end {eqnarray} \begin{eqnarray}\tag{20} \Delta \alpha &=& \sqrt {\frac {{({\alpha _ {xx}} - {\alpha _ {yy}})} ^ {2} + {({\alpha _ {yy}} - {\alpha _ {xx}})} ^ {2} + {({\alpha _ {zz}} - {\alpha _ {xx}})} ^ {2}} {2}} \end{eqnarray} \begin {eqnarray}\tag{21} \beta _ {\mathrm {tot}} &=& \sqrt {\beta _ {x} ^ {2} + \beta _ {y} ^ {2} + \beta _ {z} ^ {2}}. \end {eqnarray} Ici, \(\beta_i( i = x , y , z )\) combine les différentes quantités: \(\beta_i = (1/3) \sum_{j = x , y , z} (\beta_{ijj}+\beta_{jij}+\beta_{jji} )\).Le comportement OLN d'une molécule est généralement déterminé en comparant le moment dipolaire total (μtot ) et l'hyperpolarisabilité moyenne du premier ordre (βtot ) avec les valeurs correspondantes de l'urée [64], qui sont généralement utilisées comme valeurs seuils pour la comparaison fins. Les valeurs de μtot , α et βtot de l'urée selon nos calculs sont respectivement de 1 373 debye, 3 831 Å3 et 0,3728 × 10-30 cm5 / esu. D'après les valeurs ci-dessus, on peut voir que les valeurs μ et βtot de TTF–CH=CH–BTA dépassent celles de l'urée, ce qui corrobore l'efficacité de TTF–CH=CH–BTA en tant qu'agent OLN [65, 66, 67, 68].
Conclusions
Dans cet article, la structure moléculaire et les résultats de l'analyse vibrationnelle de la molécule TTF–CH=CH–BTA ont été rapportés avec ses propriétés électroniques. La géométrie moléculaire a été optimisée à l'état fondamental singulet, montrant un bon accord avec les données expérimentales disponibles. Les caractérisations spectrales FT-IR et FT-Raman de TTF–CH=CH–BTA sont décrites pour la première fois. Une analyse de la distribution de la charge atomique partielle a révélé une concentration de charge négative au niveau de l'atome d'azote, tandis que les atomes de carbone adjacents sont évidemment chargés positivement. Ceci est en accord avec les caractéristiques d'une surface MEP, qui montre clairement la région la plus négative autour de l'atome d'azote, car elle est très sujette à une attaque électrophile. A l'inverse, les atomes d'hydrogène apparaissent les plus chargés positivement, étant potentiellement adapté à une attaque nucléophile. Une étude de la topologie orbitale moléculaire a montré un écart d'énergie HOMO–LUMO de 2,66 eV, ce qui explique également l'origine de l'épaulement observé dans les spectres UV–Vis expérimentaux et celui calculé sur la figure 6. En particulier, les caractéristiques orbitales suggèrent qu'un transfert de charge intramoléculaire (ICT) se produit entre le donneur TTF et le fragment de benzothiazole. Les descripteurs de réactivité chimique de base ont été calculés, indiquant que la molécule TTF–CH=CH–BTA est une molécule raisonnablement molle de polarisabilité et d'activité chimique élevées. Ces résultats sont également pris en charge par les spectres UV–Vis calculés. Enfin, les propriétés optiques non linéaires calculées ont confirmé la nature de TTF–CH=CH–BTA en tant que matériau OLN potentiellement bon.
Remerciements
Ce travail a été soutenu par le MESRS-DGRSDT et l'Université de Batna 1. Les auteurs sont reconnaissants à l'Institut de Chimie de Nice, Université de Nice-Sophia Antipolis (France) pour l'utilisation de certaines ressources informatiques et l'encouragement continu.
Bibliographie
[1] F. Wudl; G. M. Smith; E. J. Hufnagel Bis-1,3=dithiolium chloride: an unusually stable organic radical cation, J. Chem. Soc. Chem. Commun., Volume 0 (1970), p. 1453-1454[2] A. Midoune; A. Messaoudi; Y. Boumedjane DFT study of a series of tetrathiafulvalene species and their redox isomer, Inorg. Chem. Commun., Volume 100 (2019), pp. 118-124
[3] M. R. Bryce Functionalised tetrathiafulvalenes: new applications as versatile ?-electronsystems in materials chemistry, J. Mater. Chem., Volume 10 (2000), pp. 589-598
[4] E. Laukhina; J. Vidal-Gancedo; V. Laukhin; J. Veciana; I. Chuev; V. Tkacheva; K. Wurst; C. Rovira Multistability in a BEDT-TTF based molecular conductor, J. Am. Chem. Soc., Volume 125 (2003), pp. 3948-3953
[5] L. Martin Molecular conductors of BEDT-TTF with tris(oxalato)metallate anions, Coord. Chem. Rev., Volume 376 (2018), pp. 277-291
[6] N. Martin; L. Sanchez; D. M. Guldi Stabilisation of charge-separated states via gain of aromaticity and planarity of the donor moiety in C\(_{{\rm 60}}\)-based dyads, Chem. Commun. (2000), p. 113-114
[7] A. Smeh; A. R. Manef DFT study of the competition between cycloaddition reactions type [2 + 2] and [4 + 2] applied to the fullerene molecule, J. Mater. Chem., Volume 5 (2014), pp. 1683-1690
[8] J. Roncali Linearly extended π-donors: when tetrathiafulvalene meets conjugated oligomers and polymers, J. Mater. Chem., Volume 7 (1997), pp. 2307-2321
[9] A. Karakas; M. Karakaya; Y. Ceylan; Y. El. Kouari; S. Taboukhat; Y. Boughaleb; Z. Sofiani Ab-initio and DFT methodologies for computing hyperpolarizabilities and susceptibilities of highly conjugated organic compounds for nonlinear optical applications, Opt. Mater., Volume 56 (2016), pp. 8-17
[10] A. Ayadi; A. Szukalski; A. K. EL Ghayoury; K. Houpa; N. Zouari; J. Mysliwiec; F. Kajzar; B. Kulyk TTF based donor-pi-acceptor dyads synthesized for OLN applications, Dyes Pigments, Volume 138 (2017), pp. 255-266
[11] T. K. Hansen; T. Jorgensen; P. C. Stein; J. Becher Crown ether derivatives of tetrathiafulvalene. 1, J. Org. Chem., Volume 57 (1992), pp. 6403-6409
[12] E. Coronado; J. R. Galan-Mascaros; C. Gimenez-Saiz; C. J. Gomez-Garcia; C. Ruis-Perez Hybrid organic/inorganic molecular materials formed by tetrathiafulvalene radicals and magnetic trimeric clusters of dimetallic oxalate-bridged complexes: the series (TTF)\(_{{\rm 4}}\){M\(^{{\rm II}}\)(H\(_{{\rm 2}}\)O)\(_{{\rm 2}}\)>[M\(^{{\rm III}}\)(ox)\(_{{\rm 3}}\)]\(_{{\rm 2}}\)} · nH2O (M\(_{{\rm 2}}\)=Mn, Fe, Co, Ni, Cu and Zn; M\(^{{\rm II}}\)=Cr and Fe; ox=C\(^{{\rm III}}\)O\(_{{\rm 2}}\)\(_{{\rm 4}}\)), J. Inorg. Chem. (2003), pp. 2290-2298
[13] H. Bengs; M. Ebert; O. Karthaus; B. Kohne; K. Praefcke; H. Ringsdorf; J. H. Wendorff; R. Wustefeld Induction and variation of discotic columnar phases through doping with electron acceptors, Adv. Mater., Volume 2 (1990), pp. 141-144
[14] M. R. Bryce; W. Devonport; L. M. Goldenberg; C. Wang Macromolecular tetrathiafulvalene chemistry, Chem. Commun., Volume 9 (1998), pp. 945-952
[15] M. Asakawa; P. R. Ashton; V. Balzani; A. Credi; C. Hamers; G. Mattersteig; M. Montalti; A. N. Shipway; N. Spencer; J. F. Stoddart A chemically and electrochemically switchable [2] catenane incorporating a tetrathiafulvalene unit A, Angew. Chem. Int. Engl., Volume 37 (1998), pp. 333-337
[16] S. Leroy-Lhez; L. Perrin; J. Baffreau; P. Hudhomme Perylenediimide derivatives in new donor–acceptor dyads, C. R. Chim., Volume 9 (2006), pp. 240-246
[17] TTF Chemistry: Fundamentals and Applications of Tetrathiafulvalene (J. Yamada; T. Sugimoto, eds.), Springer, Tokyo, 2004
[18] N. Martin; L. Sánchez; B. Illescas; I. Pérez C\(_{{\rm 60}}\)-Based electroactive organofullerenes, Chem. Rev., Volume 98 (1998), pp. 2527-2547
[19] R. M. Metzger Electrical rectification by a molecule: The advent of unimolecular electronic devices, Acc. Chem. Res., Volume 32 (1999), pp. 950-957
[20] M. Bendikov; F. Wudl; D. F. Perepichka Tetrathiafulvalenes, oligoacenenes, and their buckminsterfullerene derivatives: the brick and mortar of organic electronics, Chem. Rev., Volume 104 (2004), pp. 4891-4945
[21] A. Gorgues; P. Hudhomme; M. Sallé Highly functionalized tetrathiafulvalenes: riding along the synthetic trail from electrophilic alkynes, Chem. Rev., Volume 104 (2004), pp. 5151-5184
[22] J. L. Segura; N. Martín New concepts in tetrathiafulvalene chemistry, Angew. Chem. Int. Ed., Volume 40 (2001), pp. 1372-1409
[23] H. Fujiwara; S. Yokota; S. Hayashi; S. Takemoto; H. Matsuzaka Development of photofunctional materials using TTF derivatives containing a 1,3-benzothiazolering, Phys. B, Volume 405 (2010), p. S15-S18
[24] F. Pop; N. Avarvari Covalent non-fused tetrathiafulvalene–acceptor systems, Chem. Commun., Volume 52 (2016), pp. 7906-7927
[25] S. Yokota; K. Tsujimoto; S. Hayashi; F. Pointillart; L. Ouahab; H. Fujiwara CuII and CuI coordination complexes involving two tetrathiafulvalene-1,3-benzothiazole hybrid ligands and their radical cation salts, Inorg. Chem., Volume 52 (2013), pp. 6543-6550
[26] A. R. Dias; M. H. Garcia; P. Mendes; M. F. M. Piedade; M. T. Duarte; M. J. Calhorda; C. Mealli; W. Wenseleers; A. W. Gerbrandij; E. Goovaerts Organometallic nickel(II) complexes with substituted benzonitrile ligands. Synthesis, electrochemical studies and non-linear optical properties. The X-ray crystal structure of [Ni(\(\eta\)5-C\(_{{\rm 5}}\)H\(_{{\rm 5}}\)) {P(C\(_{{\rm 6}}\)H\(_{{\rm 5}}\))\(_{{\rm 3}}\)}(NCC6H4NH2)][PF6], J. Organomet. Chem., Volume 553 (1998), pp. 115-128
[27] S. Curreli; P. Deplano; Ch. Faulmann; A. Ienco; C. Mealli; M. L. Mercuri; L. Pilia; G. Pintus; A. Serpe; E. F. Trogu Electronic factors affecting second-order OLN properties: case study of four different push-pull bis dithiolene nickel complexes, Inorg. Chem., Volume 43 (2004), pp. 5069-5079
[28] M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; G. A. Petersson; H. Nakatsuji; X. Li; M. Caricato; A. V. Marenich; J. Bloino; B. G. Janesko; R. Gomperts; B. Mennucci; H. P. Hratchian; J. V. Ortiz; A. F. Izmaylov; J. L. Sonnenberg; D. Williams-Young; F. Ding; F. Lipparini; F. Egidi; J. Goings; B. Peng; A. Petrone; T. Henderson; D. Ranasinghe; V. G. Zakrzewski; J. Gao; N. Rega; G. Zheng; W. Liang; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; K. Throssell; J. A. Montgomery Jr; J. E. Peralta; F. Ogliaro; M. J. Bearpark; J. J. Heyd; E. N. Brothers; K. N. Kudin; V. N. Staroverov; T. A. Keith; R. Kobayashi; J. Normand; K. Raghavachari; A. P. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; J. M. Millam; M. Klene; C. Adamo; R. Cammi; J. W. Ochterski; R. L. Martin; K. Morokuma; O. Farkas; J. B. Foresman; D. J. Fox Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016
[29] A. D. Becke Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., Volume 98 (1993), pp. 5648-5652
[30] C. Lee; W. Yang; R. G. Parr Electron momentum density and x-ray structure factors of fcc-copper, Phys. Rev., Volume 37 (1988), pp. 785-789
[31] W. J. Hehre; R. F. Stewart; J. A. Pople Selfconsistent molecularorbital methods. i. use of gaussian expansions of slatertype atomic orbitals, J. Chem. Phys., Volume 51 (1969), pp. 2657-2664
[32] L. Bonniard; S. Kahlal; A. K. Diallo; C. Ornelas; Th. Roisnel; G. Manca; J. Rodrigues; J. Ruiz; D. Astruc; Jean-Yves Saillard How do nitriles compare with isoelectronic alkynyl groups in the electronic communication between iron centers bridged by phenylenebis- and -tris(nitrile) ligands? An electronic and crystal-structure study, Inorg. Chem., Volume 50 (2011), pp. 114-124
[33] P. Geerlings; F. De Proft Conceptual DFT: the chemical relevance of higher response functions, Phys. Chem. Chem. Phys., Volume 10 (2008), pp. 3028-3042
[34] P. W. Ayers; J. S. M. Anderson; L. J. Bartolotti Perturbative perspectives on the chemical reaction prediction problem, Int. J. Quantum. Chem., Volume 101 (2005), pp. 520-534
[35] J. Coates Interpretation of infrared spectra, a practical approach, Encyclopedia Anal. Chem. (2000), pp. 10815-10837
[36] R. Bozio; A. Girlando; D. Pecile Infrared and Raman spectra of TTF and TTF-d\(_{{\rm 4}}\), Chem. Phys. Lett., Volume 52 (1977), pp. 503-508
[37] D. N. Sathyanarayana Vibrational Spectroscopy-theory and Applications, New Age International (P) Limited Publishers, New Delhi, 2004
[38] M. M. Ferreira; E. Suto Atomic polar tensor transferability and atomic charges in the fluoromethane series CHxF4-x, J. Phys. Chem., Volume 96 (1992), pp. 8844-8849
[39] D. L. Beveridge; R. Lavery Theoretical Biochemistry and Molecular Biophysics: DNA. Proteins, Adenine Press, 1990
[40] P. Politzer; J. Murray The fundamental nature and role of the electrostatic potential in atoms and molecules, Theor. Chem. Acc., Volume 108 (2002), pp. 134-142
[41] B. Kosar; C. Albayrak Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol, Spectrochim. Acta A, Volume 78 (2011), pp. 160-167
[42] N. Sinha; O. Prasad; V. Narayan; A. R. Shukla Raman, FT-IR spectroscopic analysis and first-order hyperpolarisability of 3-benzoyl-5-chlorouracil by first principles, J. Mol. Simul., Volume 37 (2011), pp. 153-163
[43] G. Scalmani; M. J. Frisch; B. Mennucci; J. Tomasi; R. Cammi; V. Barone Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model, J. Chem. Phys., Volume 124 (2006), 094107 pages
[44] T. Koopmans Uber die zuordnung von wellenfunktionen und eigenwerten zu den, einzelnen elektronen eines atoms, Physica, Volume 1 (1933), pp. 104-113
[45] R. G. Parr; R. G. Pearson Absolute hardness: companion parameter to absolute electronegativity, J. Am. Chem. Soc., Volume 105 (1983), pp. 7512-7516
[46] R. J. Parr; L. V. Szentplay; S. Liu Electrophilicity index, J. Am. Chem. Soc., Volume 121 (1999), pp. 1922-1924
[47] R. G. Parr; W. Yang Density functional approach to the frontier-electron theory of chemical reactivity, J. Am. Chem. Soc., Volume 106 (1984), p. 4049-4050
[48] J. Tomasi; M. Persico Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent, Chem. Rev., Volume 94 (1994), pp. 2027-2094
[49] A. E. Reed; L. A. Curtiss; F. Weinhold Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint, Chem. Rev., Volume 88 (1988), pp. 899-926
[50] F. Weinhold; C. R. Landis Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, Cambridge, New York, Melbourr, 2005, pp. 215-274
[51] P. V. Kolinzky New materials and their characterization for photonic device applications, Opt. Eng., Volume 31 (1992), pp. 1676-1684
[52] D. F. Eaton Nonlinear optical materials, Science, Volume 253 (1991), pp. 281-287
[53] X. Sun; Q. L. Hao; W. X. Wei; Z. X. Yu; D. D. Lu; X. Wang; Y. S. Wang Experimental and density functional studies on 4-(3,4-dihydroxybenzylideneamino)antipyrine, and 4-(2,3,4-trihydroxybenzylideneamino)antipyrine, J. Mol. Struct. (Theochem), Volume 904 (2009), pp. 74-82
[54] C. Andraud; T. Brotin; C. Garcia; F. Pelle; P. Goldner; B. Bigot; A. Collet Theoretical and experimental investigations of the nonlinear optical properties of vanillin, polyenovanillin, and bisvanillin derivatives, J. Am. Chem. Soc., Volume 116 (1994), pp. 2094-2102
[55] V. M. Geskin; C. Lambert; J. L. Bredas Origin of high second- and third-order nonlinear optical response in ammonio/borato diphenylpolyene zwitterions: the remarkable role of polarized aromatic groups, J. Am. Chem. Soc., Volume 125 (2003), pp. 15651-15658
[56] M. Nakano; H. Fujita; M. Takahata; K. Yamaguchi Theoretical study on second hyperpolarizabilities of phenylacetylene dendrimer: toward an understanding of structure-property relation in nlo responses of fractal antenna dendrimers, J. Am. Chem. Soc., Volume 124 (2002), pp. 9648-9655
[57] D. Sajan; H. Joe; V. S. Jayakumar; J. Zaleski Structural and electronic contributions to hyperpolarizability in methyl p-hydroxy benzoate, J. Mol. Struct., Volume 785 (2006), pp. 43-53
[58] Y. X. Sun; Q. L. Hao; Z. X. Yu; W. X. Wei; L. D. Lu; X. Wang Experimental and density functional studies on 4-(4-cyanobenzylideneamino)antipyrine, Mol. Phys., Volume 107 (2009), pp. 223-235
[59] A. B. Ahmed; H. Feki; Y. Abid; H. Boughzala; C. Minot; A. Mlayah Crystal structure, vibrational spectra and theoretical studies of L-histidinium dihydrogen phosphate-phosphoric acid, J. Mol. Struct., Volume 920 (2009), pp. 1-7
[60] J. P. Abraham; D. Sajan; V. Shethigar; S. M. Dharmaprakash; I. Nemec; I. H. Joe; V. S. Jayakumar Efficient ?-electron conjugated push-pull nonlinear optical chromophore 1-(4-methoxyphenyl)-3-(3,4-dimethoxyphenyl)-2-propen-1-one: a vibrational spectral study, J. Mol. Struct., Volume 917 (2009), pp. 27-36
[61] S. G. Sagdinc; A. Esme Theoretical and vibrational studies of 4,5-diphenyl-2-2 oxazole propionic acid (oxaprozin), Spect. Acta. Part. A, Volume 75 (2010), pp. 1370-1376
[62] A. B. Ahmed; H. Feki; Y. Abid; H. Bonghzala; C. Minot Crystal studies, vibrational spectra and nonlinear optical properties of l-histidine chloride monohydrate, Spect. Acta. Part. A, Volume 75 (2010), pp. 293-298
[63] A. Alparone Static and dynamic electronic (hyper)polarizabilities of dimethylnaphthalene isomers: characterization of spatial contributions by density analysis, Sci. World J., Volume 2013 (2013), 832682 pages
[64] Y. X. Sun; Q. L. Hao; W. X. Wei; Z. X. Yu; L. D. Lu; X. Wang; Y. S. Wang Experimental and density functional studies on 4-(3,4-dihydroxybenzylideneamino) antipyrine, and 4-(2,3,4-trihydroxybenzylideneamino)anti-pyrine, J. Mol. Struct.: THEOCHEM, Volume 904 (2009), pp. 74-82
[65] A. I. de Lucas; N. Martin; L. Sfinchez; C. Seoane; R. Andreu; J. Garin; J. Orduna; R. Alcalfi; B. Villacampa The first tetrathiafulvalene derivatives exhibiting second-order OLN properties, Tetrahedron, Volume 54 (1998), pp. 4655-4662
[66] M. Gonzalez; J. L. Segura; C. Seoane; N. Martin Tetrathiafulvalene derivatives as OLN-phores: synthesis, electrochemistry, Raman spectroscopy, theoretical calculations, and OLN properties of novel TTF-derived donor-\(ð\)>-acceptor dyads, J. Org. Chem., Volume 66 (2001), pp. 8872-8882
[67] A. Szukalski; A. Ayadi; K. Haupa; A. El Ghayoury; B. Sahraoui; J. Mysliwiec All-optical switching and two-states light controlled coherent-incoherent random lasing in thiophene based donor acceptor, Chem. Phys. Chem., Volume 19 (2018), pp. 1605-1616
[68] A. Colombo; C. Dragonetti; D. Marinotto; S. Righetto; G. Griffini; S. Turri; H. Akdas-Kilig; J. Fillaut; A. Amar; A. Boucekkinee; C. Katan Nonlinear optical properties of intriguing Ru σ-acetylide complexes and the use of a photocrosslinked polymer as a springboard to obtain SHG active thin films, Dalton Trans., Volume 45 (2016), pp. 11052-11060